Selektívna insuficiencia IgA je najbežnejšia primárna imunodeficiency stav (PIDS). Schopnosť pacientov s selektívnym nedostatkom IgA sa pohybuje od 1: 400 do 1: 1000 v európskej populácii a výrazne nižšie, od 1: 4000 do 1: 20 000, v mongoloid. V Spojených štátoch sa prevalencia ochorenia pohybuje od 1 do 223-1000 v skupine, ktorá študovala až 1 až 400-3000 u zdravých darcov krvi. V Rusku sa takéto štúdie neuskutočnili.
Pre tento stav sa selektívny pokles sérovej koncentrácie IgA charakterizuje pod 0,05 g / l (u detí starších ako štyri roky) normálna úroveň iné sérové \u200b\u200bimunoglobulíny, normálna reakcia Sérové \u200b\u200bprotilátky a konvenčná bunka-nepriama imunitná reakcia. Vo väčšine štúdií, frekvencia výskytu medzi mužmi a ženskými predstaviteľmi bola približne rovnaká.
Ľudia s neschopnosťou produkovať IGA môžu niesť svoju chorobu asympticky v dôsledku kompenzačných mechanizmov alebo trpia častými respiračnými infekciami, tráviacimi alebo dobrý systém, gastroenterologická patológia (napríklad celiakova choroba), tendenciu k atopickým poruchám, ako je polyniesis, bronchiálna astma, Atopická dermatitída, igé-nepriame potravinová alergia, rovnako ako neurologické a autoimunitné ochorenia (najčastejšie je to reumatoidná artritída, systémová red lupus, idiopatická trombocytopenická fialová, shegreen syndróm). S selektívnou insuficienciou IGA alergické ochorenia, ako je atopická dermatitída a bronchiálna astma, sa stretli v 40% prípadov (Consilium Medicum, 2006). Tiež charakteristické pre väčšinu takýchto pacientov anafylaktických reakcií pri preplnení zložiek krvi a podávanie intravenóznych imunoglobulínov, ktoré sú spojené s prítomnosťou IgA v týchto produktoch.
Klinické príznaky selektívneho nedostatku IgA sa môžu prejaviť v ranom detstve, pričom vek rovnakej frekvencie a závažnosti prenosných infekcií sa môže znížiť v dôsledku kompenzačného zvýšenia protilátok sub-stupňov IgG1 a G3, IgM. Ďalšie vysvetlenie neprítomnosti klinické príznaky Môže existovať normálna úroveň sekrečnej IGA, napriek zníženiu hladiny sérového imunoglobulínu. Alebo naopak, niektorí pacienti s pôvodne diagnostikovaným deficite IgA môžu vyvinúť kliniku so spoločným variabilným imunitným zlyhaním.
Terapia selektívneho nedostatku IgA je v súčasnosti pri identifikácii súbežných ochorení preventívny mérpruhy Znížiť riziko infekcie, ako aj rýchlu a účinnú liečbu infekcií.
Neexistuje žiadna špecifická liečba. Predpoveď u pacientov s nedostatkom IgA je všeobecne dobrá, ak nie je vyjadrená klinické prejavy. Insuficiencie IgA u detí sa môže časom kŕmiť.
Byť geneticky stanovený, stavy imunodeficiencie vznikajú v dôsledku defektov genetického zariadenia. Pacienti so spoločným variabilným imunitným zlyhaním a selektívnym nedostatkom IgA sa často detegujú v jednej rodine a majú spoločný HLA-haplotyp; Mnohí majú vzácne alely a génové delécie vo vnútri triedy 3 triedy 3 na chromozóme 6. Nedávno bolo preukázané, že niektoré rodinné prípady celkového variabilného imunitného zlyhania a selektívneho deficitu IgA sú spôsobené mutáciou génu TNFRSF13B, ktorá kóduje proteín známy ako TACI (transmembránový aktivátor a interaktor vápnika a cyklofilín-ligand). Je pravdepodobné, že v prípadoch, keď neboli zistené TACI mutácie, príčina chorôb by mohla slúžiť ako spontánne alebo dedičné mutácie iných génov, ešte nie sú upevnené.
V súčasnej dobe, možné klinické prejavy selektívneho deficitu IgA, možnosti prúdenia, možných súbežných ochorení sú podrobne opísané. Rozhodujúcim v diagnostike ochorenia je selektívny pokles koncentrácie séra IgA u detí od 4 rokov pod 0,05 g / l na normálnej úrovni iných sérových imunoglobulínov v opakovaných imunogramoch. Liečba sa skladá z identifikácie súbežných ochorení, ktoré vykonávajú preventívne opatrenia na zníženie rizika infekcie a je tiež potrebné rýchlo a Účinná liečba infekčné choroby.
Informácie o frekvencii výskytu tejto primárnej imunodeficiencie štátu v ruskej populácii chýba, čo neumožňuje porovnať prevalenciu choroby v našej krajine s inými krajinami, ak sa takéto štúdie už uskutočnili.
Hlavným problémom je absencia jednotných odporúčaní o taktike pacientov s selektívnym nedostatkom IGA.
S cieľom posúdiť frekvenciu výskytu selektívneho nedostatku IgA medzi deťmi skupiny dávkovacie pozorovanie "Často choré deti" a charakterizujú spektrum svojich klinických prejavov Ruská federácia Na základe FGBU "FNCC Dgoo pomenoval po Dmitriji Rogachev" Ministerstvo zdravia Ruskej federácie a GBUZ DGKB č. G. N. Speransky Disk bol vykonaný túto prácu.
Metódy materiálov a výskumu
Deti so selektívnym nedostatkom IgA, pozorované v GBUZ DGKB č. 9 sa stali predmetom štúdie. G. N. Speransky disk. Okrem toho retrospektívne analyzoval lekárske záznamy na obdobie rokov 2003 až 2010. 9154 pacientov zo skupiny dávkovacieho pozorovania "často choré deti" (tabuľka 1-3).
Počas skúšky sa použili tieto metódy:
- klinické a anamnestické;
- všeobecne I. biochemické analýzy krv;
- imunologická štúdia zloženia krvi spôsobmi olejovitou a prietokovou cytometriou;
- vzorky na skarifikáciu;
- stanovenie špecifickej metódy IgE imunoblotovania;
- Štúdium funkcie vonkajšieho dýchania;
- rinokitologický výskum.
Diagnóza selektívneho nedostatku IgA bola zvýšená na základe selektívneho zníženia sérovej koncentrácie IgA pod 0,05 g / l normálne ukazovatele Ostatné sérové \u200b\u200bimunoglobulíny v opakovaných imunogramoch a vylúčenie iných možné príčiny ich nedostatočnosť pre deti nad 4 rokov.
Pri zhromažďovaní anamnézy osobitná pozornosť Platená frekvencia a spektrum klinických prejavov, \\ t súbežná patológia, ako aj rodinná história študovala podrobne. Klinické vyšetrenie detí sa uskutočnilo v súlade so všeobecne akceptovanými metódami. Obsah imunoglobulínov tried A, G, M, E v sére bol určený metódou merača oleja na merači BN 100 (Dade Bering, Nemecko) pomocou Dade Behring Set. Fenotypovanie lymfocytov sa uskutočnilo metódou prietokovej cytometrie na zariadení FACSSCAN (Becton Dickesson, USA) s použitím fluorescenčne označených monoklonálnych protilátok sú súčasne (Becton Dickesson, USA). Pacientov s akýmikoľvek prejavmi atopie, ako aj všetkým pacientom zvýšená úroveň IgE, ktorá bola identifikovaná v dôsledku odhadovaných ukazovateľov imunitného stavu metódou merača oleja, bola vykonaná alergologická závislosť metódou testov skarifikácie u detí starších ako 4 roky alebo stanovením špecifickej IgA v sére u pacientov pod 4 4 rokov. Deti s diagnózou "bronchiálnej astmy" alebo prítomnosť broncho-prestaveného syndrómu v histórii uskutočnili štúdiu funkcie vonkajšieho dýchania na prístroji Spirovit SP-1 (Schiller AG, Švajčiarsko). Všetky potrebné adresy a konzultácie s príbuznými špecialistami, pričom sa zohľadnia dostupné sťažnosti.
Výsledky a jeho diskusiu
Retrospektívna analýza lekárskych záznamov pacientov s vodiacimi diagnózami "recurient ARVI", "CHBD", "CBD", ako aj "EBD" umožnil stanoviť, že frekvencia selektívneho nedostatku IgA v tejto skupine detí je dva a dokonca trikrát vyššia ako v populácii.
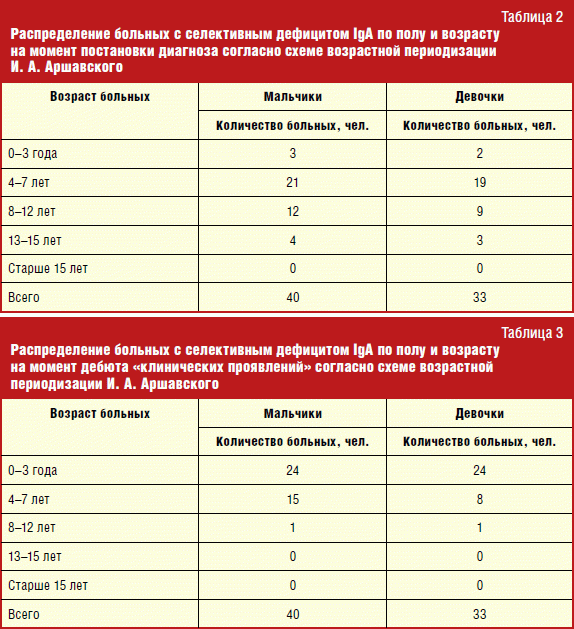
Absolútne číslo, ako aj percento detí s touto primárnou imunodeficienciou, možno vidieť v tabuľke. štyri.
Bohužiaľ, údaje za rok 2007 nie sú k dispozícii. V rokoch 2003 a 2004 Konzultovalo sa 692 a 998 detí. Medzi nimi boli celkom 5 pacientov so selektívnym nedostatkom IgA, čo je o niečo častejšie ako v priemere populácie - 1: 346 a 1: 333, proti 1: 400-600. Od roku 2005 sa frekvencia prvýkrát identifikovala pacientov s týmto PID dramaticky: 1: 113 v roku 2005, 1: 167 v roku 2006, 1: 124 v roku 2008, 1: 119 v roku 2009 a nakoniec, 1: 131 v roku 2010. Počas štúdie sa frekvencia výskytu zmenila z 1: 346 v roku 2003 na 1: 131 v roku 2010, keď sa ukázalo byť v porovnaní s predchádzajúcimi rokmi. Zvýšenie frekvencie výskytu pacientov so selektívnym nedostatkom IgA na tretí rok po začatí práce by mala byť spojená so zvýšenou bdelosť lekárov vo vzťahu k tejto patológii, ako aj zlepšenie laboratórne diagnostika. Je potrebné pokračovať v rozšírení vedomostí o lekárov o tejto chorobe, pretože tok detí, ktoré rodičia vedú k imunológu sťažnosťami Časté choroby, Zvýšenie z roka na rok.
V priebehu tejto práce bolo aj 235 detí a 32 dospelých aj prospektívne.
Hlavnou skupinou bola 73 detí s diagnostikovaným selektívnym nedostatkom IGA.
Druhá skupina pacientov zahŕňala 153 detí s idiopatickou trombocytopenics fialová (ITP). Posúdenie imunitného stavu pacientom s ITP sa uskutočnilo s cieľom identifikovať selektívny nedostatok IgA medzi nimi, pretože táto korelácia bola opísaná vo svetovej literatúre a rovnaké údaje boli získané počas táto štúdia. Nie je to jedno dieťa s nedostatkom IgA medzi nimi nebola identifikovaná. Napriek tomu, že počas preskúmania imunitného stavu pacientov s ITP, nemohli sme identifikovať selektívny nedostatok IgA medzi nimi, iné malé humorálne chyby boli identifikované: nedostatok podtriedy IgG, detský hypogammaglobulinémia, čiastočný pokles IgA.
Tretia skupina zahŕňala 32 dospelých vo veku od 20 do 54 rokov, ako aj 8 detí vo veku od 4 do 10 rokov, ktoré sú najbližšími príbuznými pacientmi s selektívnym nedostatkom IgA, ktoré sa hodnotí imunitný stav, aby hľadal a opísal rodinné prípady .
Počas prieskumu a analýzy získaných údajov boli získané výsledky opísané nižšie.
Pomer muža a ženy u pacientov s selektívnym nedostatkom IgA bol približne rovnaký. Preskúmalo sa 40 chlapcov a 33 dievčat. To zodpovedá svetovej literatúre.
Vrchol detektívnosti selektívneho nedostatku IGA predstavoval vo veku 4-7 rokov. Opakované infekčné ochorenia, spravidla vzniknuté nízky vek Alebo so začiatkom návštevy detskej predškolskej inštitúcie. Spravidla, predtým, ako sa dostanete k imunológovi, deti nahromadili určitú infekčnú históriu, pretože existujú určité príznaky, ktoré im umožňujú podozrenie na PID. A okrem toho, aj keď štúdia bola vykonaná v skoršom veku a absencia IgA bola odhalená do 4 rokov, nedovolila nám, aby sme dali jednoznačnú diagnózu PID, nemohli sme úplne vylúčiť nezrelosť systému syntézy imunoglobulínov. Preto bola diagnóza vystavená až 4 roky na otázky a odporúčané pozorovanie v dynamike. Z tohto dôvodu interval 4-7 rokov.
Vedúce sťažnosti pri manipulácii s deťmi s selektívnym nedostatkom IgA boli časté respiračné vírusové infekcie s nekomplikovaným tokom. Debetné opakujúce sa respiračné ochorenia, spravidla účtované vo veku do 3 rokov. Zodpovedá tiež svetovej literatúre. Keďže dynamická kontrola nad väčšinou pacientov našej štúdie bola vykonaná na dlhú dobu, niekoľko rokov, niekedy pred prechodom pacienta v sieti pre dospelých, možno argumentovať, že s vekom sa znížila frekvencia a závažnosť prenosných infekcií. Predpokladalo sa, že to bolo spôsobené kompenzačným zvýšením protilátok subtrysieho IgG1 a IgG3, IGM, ale táto otázka si vyžaduje ďalšiu štúdiu. Druhá frekvencia je sťažnosť pri aplikácii, bola častá Orvis tečie s komplikáciami. Znížila sa frekvencia komplikovaného, \u200b\u200batypicky, ktorá postupovala ORVI s vekom u našich pacientov, ako je znázornené dynamické pozorovanie.
Z spektra infekčných chorôb u pacientov so selektívnym nedostatkom IGA, popredné miesto obsadili infekčnými chorobami ENT orgánov a nižších infekcií dýchacie cesty. Je to spôsobené tým, že pokles sekrečnej IGA, ktorý je súčasťou miestnej imunity, vedie k ľahkej infekcii a reprodukcii mikroorganizmov na slizničných membránach, najzraniteľnejších voči kontaktom infekčné chorobyprenášanie vzduchových kvapiek.
V spektre nekomuničných ochorení sa zjavná korelácia odhalila s autoimunitnými ochoreniami, ktoré sú najdôležitejšie prejavy selektívneho nedostatku IgA, najmä s idiopatickou trombocytopenickou fialovou činnosťou (1,5-2 na 100 tisíc).
Z autoimunitné ochorenia U pacientov s selektívnym nedostatkom IgA, juvenilnej reumatoidnej artritídy (4-krát), chronickej idiopatickej trombocytopenickej fialovej fialovej (3-krát), autoimunitnej hepatitídy (3-krát) boli najčastejšie. Okrem toho, podľa svetovej literatúry, u pacientov s selektívnym nedostatkom IgA, zvýšená frekvencia autoimunitných štátov medzi najbližšími príbuznými. Podľa nášho výskumu ich počet však neprekročila hodnoty zovšeobecnenia.
Frekvencia atopických ochorení u pacientov s selektívnym nedostatkom IgA sa ukázala byť výrazne vyššia ako v populácii (tabuľka 4). Pre všeobecný jazyk je porovnateľný len s frekvenciou alergickej rinitídy. Takéto pripomienky sa odrážajú v mnohých predtým uskutočnených štúdiách. Nemožno povedať, že alergické ochorenia u väčšiny pacientov s nedostatkom IgA pokračujú ťažko ako u ľudí bez tohto imunologického defektu. Väčšia prevaha atopie však dáva dôvod na zvýšenie otázky vykonávania imunologického vyšetrenia s cieľom identifikovať formy selektívneho nedostatku IgA, ktoré sa zatiaľ nepreukázali. Aj keď to nemusí mať rozhodujúcu úlohu, pokiaľ ide o prístup k terapii súčasného atopického stavu, ale pomôžem urobiť diagnózu včas a zistí sa možné riziká pre ľudí, ktorí majú selektívny nedostatok IgA.
Pri analýze opakovaných imunogramov počas dynamického pozorovania u detí s selektívnym nedostatkom IgA, v súvislosti s perzistentnými zmenami laboratórnych ukazovateľov, boli pridelené dva veľké skupiny pacientov. V skupine A, nebola žiadna IGA bez ďalších zmien. Skupina v neprítomnosti IgA v kombinácii s perzistentným zvýšením IgG. Vykonala sa porovnávacia analýza týchto skupín pacientov.

Vek debut klinických prejavov v týchto skupinách nebol spoľahlivo odlišný.
Bolo zistené, že u pacientov s selektívnym nedostatkom IgA, zvýšenie hladiny IgG koreluje s opakovanými infekčnými ochoreniami pokožky a mäkkých tkanív. Táto otázka si vyžaduje ďalšie štúdium.
Pri porovnaní týchto skupín pacientov neboli zistené významné rozdiely v spektre alergopatológie.
V priebehu práce sa uskutočnilo hodnotenie imunitného stavu v 20 rodinách pacientov s selektívnym nedostatkom IgA. 4 Rodinné prípady boli odhalené. Okrem toho sa zhromaždila podrobná rodinná história. Medzi dospelými príbuznými s zaťaženou infekčnou históriou, ktorá sa podarilo vykonať prieskum, boli tieto alebo iné poškodenie humorálnej imunity. V súlade s tým, pri identifikácii malých humorálnych defektov (najmä selektívnym nedostatkom IgA), je povinné preskúmanie najbližších príbuzných, najmä v prítomnosti zaťaženej infekčnej anamnézy.
Vzhľadom k tomu, že selektívny nedostatok IGA medzi deťmi vydávajúcej pozorovacej skupiny "často chorých detí" je oveľa častejšie ako v spoločnej populácii detí, praktizovanie pediatrovia musia byť opatrní v porovnaní s týmto ochorením. Nie je vždy ľahké podozrenie, pretože klinické prejavy sú veľmi variabilné: z asymptomatických foriem na opakované bakteriálne infekcie s potrebou častého antibakteriálna terapia. Odporúča sa rozšíriť vedomosti o pediatroch a úzkych ambulantných a stacionárnych špecialistov o malých dedinkách humorálnej imunity.
Vzhľadom k tomu, u pacientov s selektívnym nedostatkom IgA sú významne vyššie ako frekvencia alergickej patológie (bronchiálna astma, atopická dermatitída, potravinové alergie), nad frekvenciou autoimunitných ochorení a hematologických ochorení, ako aj frekvencie chronické choroby (ENT orgány, urogenitálny systém, gastrointestinálny trakt) ako v populácii je jeho identifikácia povinná na účely poskytovania plnohodnotných a jeho dočasných zdravotná starostlivosť Pacientov.
Odporúča sa, aby sa odporúča konzultovať s imunológom / imunologickým vyšetrením detí s hydratovanou infekčnou anamnezúrou, pacientov s hematologickými a autoimunitnými ochoreniami, aby vykonali skríningové prieskumy úrovne všeobecných pacientov s IgA s prítomnosťou alergických ochorení.
V priebehu štúdie sa zistilo, že väčšina detí s selektívnym deficitom IgA označila koreláciu autoimunitná patológia a perzistentný nárast IgG v opakovaných imunogramoch. S inými ochoreniami takáto korelácia nebola stanovená. Podobné zmeny ukazovateľov sú rizikovým faktorom v autoimunitnej patológii dieťaťa a vyžadujú osobitnú pozornosť.
Napriek tomu, že korelácie medzi prítomnosťou rodinnej histórie selektívneho nedostatku IgA a závažnosti klinických prejavov u pacientov nebola stanovená, pre týchto pacientov, vyšetrenie najbližších príbuzných, najmä v prítomnosti zaťaženej infekčnej anamnézy , Je povinné.
Literatúra
- Hammarstrom L., LonnQVist B., Ringden O., Smith C. I., WIEBE T. Prenos deficientu IgA na pacienta s kostnou dreňou s aplastickou anémiou // lancetou. 1985; 1 (8432): 778-781.
- Latiff A. H., KERR M. A.Klinický význam imunoglobulínu Deficit // Annals klinickej biochémie. 2007; 44 (PT 2): 131-139.
- Al-ats R. A., RAHI A. H.Nedostatok primárnych protilátok v Araboch: prvá správa z východnej Saudskej Arábie // Journal of Clinical Immunology. 1998; 18 (5): 368-371.
- Carneiro-Sampaio M. M., karbonare S. B., Rozentraub R. B., De Araujo M. N., Riberiro M. A., Porto M. H.Frekvencia selektívneho deficitu IgA medzi brazílskymi darcami krvi a zdravými tehotnými ženami // alergologickým imunopatológiou (MADR). 1989; 17 (4): 213-216.
- EZEOKEOK A. C. Selektívny nedostatok IgA (SIGAD) vo východnej Nigérii // African Journal of Medicine a Medical Sciences. 1988; 17 (1): 17-21.
- Feng L. Epidemiologická štúdia selektívneho deficitu IgA medzi 6 národnosť v Číne // Zhonghua Yi Xue ZA ZHI. 1992; 72 (2): 88-90, 128.
- Pereira L. F., Sapina A. M., Arroyo J., Vinuelas J., Bardaji R. M., Prieto L. Prevalencia selektívneho nedostatku IgA v Španielsku: Viac ako my ho // krv. 1997; 90 (2): 893.
- Wiebe V., Heal A., Lefranc M. P., Lefranc G. Molekulárna analýza T17 Immunoglobulín CH multigénu delécie (del A1-GP-G2-G4-E) // ľudská genetika. 1994; 93 (5): 5.
L. A. Fedorova *,
E. S. PUSKKOVA *
I. A. KORSUNSKY **, 1, Kandidát na lekárske vedy
A. P. PRODEUS *,doktor lekárskych vied, profesor
Imunodeficiencie - znížené kvantitatívne ukazovatele a / alebo funkčná aktivita hlavných zložiek imunitný systém, čo vedie k porušeniu ochrany tela z patogénnych mikroorganizmov a prejavuje sa zvýšeným incidenciou.
Ako je známe, hlavnou funkciou imunity systému je rozpoznávanie a eliminácia cudzích látok antigénnej povahy, ktorá preniká do tela okolitý (mikroorganizmy) alebo endogénne vznikajúce (nádorové bunky). Táto funkcia je implementovaná pomocou faktorov. vrodená imunita (fagocytóza, antimikrobiálne peptidy, proteíny komplementu systému, NK bunkových systémov atď.) A získané, alebo adaptívne imunitné, uskutočňované s použitím bunkových a humorálnych imunitných reakcií. Regulácia aktivity zložiek organizmu imunitnej ochrany a ich interakcia sa vyskytuje s pomocou cytokínov a intercelulárnych kontaktov.
V každej z uvedených zložiek imunitného systému, ako aj v mechanizmoch ich regulácie, môžu dôjsť k poruchám, čo vedie k rozvoju imunodeficiencie, ktorého hlavným klinickým prejavom je zvýšená citlivosť na kauzálny činidlá infekčných ochorení. Dva typy imunodeficiencie rozlišujú: primárne a sekundárne.
Primárna imunodeficiencia(Pid) - dedičné ochoreniaspôsobené vadami génov, ktoré kontrolujú imunitnú reakciu. PID - choroby, rôznorodé v povahe a závažnosti imunitných defektov, klinických prejavov a molekulárnych porúch. Pre klinický obraz je PID charakterizovaná opakovanými a chronickými, závažnými infekčnými procesmi, do väčšieho stupňa bronchopulmonálneho systému.
a engál, kožné a sliznice; Môžu sa vyvinúť hnisavé lymfadentes, abscesy, osteomyelitída, meningitída a sepsa. V niektorých formách existujú prejavy alergií, autoimunitných ochorení a vývoj niektorých malígne nádory. Mali by ste venovať pozornosť vekové ukazovatele fyzický vývoj. V súčasnosti je opísaných približne 80 PID, gény zodpovedné za rozvoj väčšiny týchto chorôb. Primerané laboratórne testy umožňujú diferencovať patológiu na úrovni lymfocytov a patológie na úrovni neebulárnej deštrukcie a mechanizmov odstraňovania antigénu.
PID Prevalenciazávisí od formy ochorenia a v priemere od 1: 10 000 do 1: 100 000 novorodencov. Selektívny deficit IgA napríklad nastáva oveľa častejšie od 1: 500 do 1: 1500 ľudí všeobecnej populácie. Prevaha rôzne tvary Pid sa líši rozdielne krajiny. Najčastejšie sú vady protilátok - 50-60% prípadov, kombinované PID - 10-30%, defekty fagocytózy - 10-20%, defekty komplementu - 1-6%. Väčšina PID sa prejavuje v ranom detstve, hoci je možné lepšie byť začiatkom niektorých foriem PID, najmä celkového variabilného imunologického zlyhania (ovin).
V prípade mechanizmov vývoja sa prideľujú 4 hlavné skupiny PID:
1. skupina - prevažne humorálna alebo v bunke
Pid;
2. Group - kombinovaná PID (so všetkými imunodeficatormi T-buniek Existuje porušenie funkcie B buniek);
3RD Group - PID, v dôsledku defektov fagocytózy;
4. Skupina - PID, vďaka vadom v systéme komplementu.
Princípy diagnostiky primárnej imunodeficiencie
Včasná diagnóza a včasný začiatok liečby určujú prognózu ochorenia. Formulácia diagnózy na úrovni okresných pediatroch prezentuje určité ťažkosti, čo je často spôsobené absenciou možnosti včasného poradenstva s pacientom s lekárskym imunológom a vykonávaním špeciálneho laboratórneho imunologického vyšetrenia (tabuľka 11-1). Hoci znalosť charakteristík PID klinického obrazu a zmeny
vo všeobecných klinických laboratórnych testoch vám umožňuje podozrenie na PID a poslať pacienta odborníkom v odbore. Európska spoločnosť imunodeficiencie spoločnosť vyvinula protokoly na včasnú diagnostiku PID, a tiež vytvorená elektronická základňa Údaje Európskeho registra PID. PID diagnostický algoritmus je znázornený na obr. 11-1.
Tabuľka 11-1.Etapy imunologického vyšetrenia s podozrením na imunodeficienciu
Obr. 11-1.Algoritmus pre diagnostiku primárnej imunodeficiencie
Všeobecné vlastnosti klinického obrazu primárnej imunodeficiencie
Vedúci B. klinický obraz PID je tzv. infekčný syndróm - zvýšená citlivosť na kauzálnych látok infekčných chorôb vo všeobecnosti, nezvyčajne prísne recidivujúce (opakované) klinický prúd, Prítomnosť v etiológii ochorenia atypických patogénov (často oportunistic). Typ patogénu je určený povahou imunitnej defektu. S protilátkovými defektmi je možné identifikovať stabilné antibakteriálne lieky Posuvník - Staphylococci, Streptococci, Hemofilic Wand. S imunitným zlyhaním T-buniek, okrem baktérií, vírusy odhaľujú (napríklad rodina herpesvírusov), huby (Candida spp., Aspergillusa kol.) A s fagocytovým defektom - Staphylococci, gram-negatívne baktérie, huby atď.
Laboratórny výskum
Ak vám klinické údaje umožňujú podozrenie na PID, mali by sa vykonať tieto štúdie: \\ t
Stanovenie nasadeného vzorca krvi (najmä kvantitatívne a percentuálne ukazovatele lymfocytov);
Definícia hladín IgG, IgA a IgM v krvnom sére;
Výpočet subpopulácií T- a B-lymfocytov;
◊ Analýza funkčného stavu fagocytov (najjednoduchšia a informatívna analýza je test na obnovu tetrazolium blue);
◊ Analýza obsahu hlavných zložiek komplementu (začiatok s C3 a C4);
◊ Analýza pre infekciu HIV (ak je to možné rizikové faktory);
◊ Molekulárne genetické štúdie, keď svedectvo.
Princípy liečby primárnej imunodeficiencie
Hlavným cieľom PID terapie je liečba komplikácií ochorenia a ich prevencie. Tento prístup je spôsobený tým, že vady imunitného systému s PID sú položené na genetickej úrovni. V súčasnosti vykonáva intenzívny výskum na géne
Šluková terapia imunodeficiencie, ktorá môže viesť k vzniku radikálnejších metód ich liečby.
V závislosti od formy PID je liečba vykonávať substitučnú terapiu, liečbu a prevenciu infekčných, autoimunitných prejavov ochorenia, liečby malígnych neoplazmy a aplikácie ŠPECIÁLNE METÓDYvrátane transplantácie hematopoetických kmeňových buniek (v závislosti od typu PID).
Defekty imunoglobulínu
Prechodová hypogammaglobulinémia u detí
Prechodná hypogammaglobulinémia u detí je spojená s fyziologickým znakom fázovanej tvorby imunoglobulínového systému. Zrenie IgM a IgA protilátok je najviac "oneskorenia". U zdravých detí sa obsah materského IgG postupne znižuje a po pol roka zvyšuje vývoj vlastných IgG protilátok. V niektorých deťoch sa však zvýšenie úrovne imunoglobulínov oneskorí. Takéto deti môžu trpieť opakovanými bakteriálnymi infekčnými ochoreniami. V týchto prípadoch by sa nemalo uchýliť k infúziám liekov darcovských imunoglobulínov (zavedenie intravenózneho imunoglobulínu).
Selektívny imunoglobulínový nedostatok A
Selektívny imunoglobulín A (SD IGA - Selektívny nedostatok IGA)v dôsledku defektu génu tnfrsf13b.
alebo p). Deficit IgA v prítomnosti imunoglobulínov iných tried je najčastejšou imunodeficienciou zistenou v spoločnej populácii s frekvenciou 1: 500-1500 ľudí (u pacientov trpiacich alergiami, častejšie). IGA Selektívna insuficiencia, t.j. Prispievanie v nedostatku jednej zo podtried (30% prípadov) a kompletné (70% prípadov). Nedostatok podtriedy IGA2 vedie k výraznejšiemu klinickému obrazu ako deficit podtriedy IGA1. Kombinácie nedostatku IgA s inými porušeniami sú možné: s defektom IgG biosyntézy a anomálie T-lymfocytov. Prevažná väčšina osôb s selektívnym
deficit IgA je prakticky zdravý. Pre deti do 2 rokov je nedostatok IgA fyziologickým stavom.
Odhaliť zníženie koncentrácie séry IgA<5 мг/дл у детей старше 4 лет; IgG и IgM в норме, количество и соотношение субпопуляций лимфоцитов и их функциональная активность могут быть в норме.
Klinický obraz.S nedostatkom IgA, 3 skupiny patologických syndrómov sa môžu vyvinúť: infekčné, autoimunitné a alergické. Pacienti s nedostatkom IgA sú predisponované rekurentným infekčným ochoreniam horných dýchacích ciest a tráviacich orgánov. Najčastejšie a vážne tečie sú rôzne autoimunitné ochorenia (reumatoidná artritída, ankylozujúca spondylitída, Shegreen syndróm, vaskulitída s léziou mozgových ciev, autoimunitná tyreoiditída, SLE, glomerulonefritída, hemolytická anémia, diabetes typu I, vitiligo atď.). Frekvencia celiachood presiahne tak u detí s normálnou IGA 10-krát. Najčastejšie zistené alergické prejavy: neznášanlivosť k proteínu kravského mlieka, atopickej dermatitídy (ATD), bronchiálnej astmy.
Liečba.Prípady asymptomatického prietoku nevyžadujú žiadne špeciálne ošetrenie; V prítomnosti klinických prejavov infekčných, autoimunitných a alergických ochorení sa liečba vykonáva v súlade s normami.
Náhradná terapia imunoglobulínov darcov nie je znázornená v selektívnom, ani s úplnou nedostatkom IgA, pretože pravdepodobnosť tvorby anti-etehtiotických protilátok proti IGA a vývoj transfúznych komplikácií spôsobených nimi je vysoká.
Agamaglobulinémia s nedostatkom B-buniek
X-Cutched Aghamaglobulinémia (brutonova choroba)je to 90% všetkých prípadov AGHAMAGLOBULINEMIA. Chlapci sú chorí, synovia (אּ, ρ) nosičov chybného génu bTK (XQ21.3-Q22),b-lymfocytovo-špecifická proteindhyroskinkináza BTK (Brutonová tyrozínková kináza- Bruton Tyrozínkináza). V dôsledku vady, porušenie intracelulárnych signalizačných dráh, rekombinácie ťažkých imunoglobulínových reťazcov,
obnovenie trans B-buniek v B-lymfocytoch. U 10% pacientov s nedostatkom AGAMMAGLOBULINEmiA s nedostatkom v bunke, autosomonéza je zdedená. V súčasnosti je opísaných 6 genetických defektov, vrátane molekúl receptorov pred B-buniek, cytoplazmatického adaptérového proteínu B-buniek (BLNK) a génu Opakovanie bohatý na leucín (LRRC8).
Laboratórne údaje.Neexistujú žiadne periférne B-lymfocyty. V kostnej dreni sú pred-B bunky s μ-reťazcom v cytoplazme. Počet T-lymfocytov a funkčných testov na T-lymfocytoch môže byť normálne. IgM a IgA v krvi nemožno odhaliť; IgG môže byť prítomný, ale v malých množstvách (0,4-1,0 g / l). Žiadne protilátky proti krvným skupinám antigény a vakcínové antigény (tetanus, záškrtheria toxíny atď.). Neutropénia sa môže vyvinúť. Histologická štúdia lymfoidnej tkaniny: V lymfoidných folikulách nie sú žiadne zárodočné (zárodočné) centrá a plazmatické bunky.
Klinický obraz.Ak je rodinná história neznáma, diagnóza sa v priemere zrejmé ako 3,5 roka. Choroba sa vyznačuje lymfoidným tkanivom hypoplasia, silne tečúcou hnisavými infekciami, infekčnými ochoreniami horných (sinusites, otitis) a nižšia (bronchitída, pneumónia) dýchacích ciest; Gastroenteritída, pyodermia, septická artritída (bakteriálna alebo chlamydious), septikémia, meningitída, encefalitída, osteomyelitída. Ako kauzatívne činidlá respiračných ochorení, najčastejšie vykonávané Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus,hnačka črevnej baktérie alebo giardia Giardia Lamblia.Pacienti s Aghamaglobulinémiou sú tiež citlivé na infekčné ochorenia spôsobené mykoplazmom a uraplazmi, ktoré spôsobujú vývoj chronických pneumónia, hnisavých artritídy, cystitu a subkutánneho abscesu. Z vírusov typických neurotropných vírusov ECHO-19 a kokes, čo spôsobuje ťažkú \u200b\u200bostrú a chronickú encefalitídu a encefalomyelitídu. Prejavy enterovírusových infekcií môžu byť syndróm ako dermatomy, ataxia, bolesti hlavy, porušovanie správania. U pacientov s deťmi v imunizácii, živý polyovakcinát, spravidla, dlhodobo uvoľňovanie vírusu POLOŽE sa deteguje prostredníctvom slizníc a s obnovenou a zvyšujúcou sa virulenciou (t.j. v detskom kolektore
sme skutočným nebezpečenstvom infekcie zdravých detí s POLOHA, v dôsledku kontaktu s očkovaným imunodeficientným dieťaťom). Autoimunitné poruchy počas agamaglobulinémie môžu byť reprezentované reumatoidnou artritídou, syndrómou podobným sklerermovým, scrugarl, nešpecifický ulcerózna kolitída, diabetes typu I (kvôli prevahu thl imunitnej reakcie).
Fyzickú kontrolu.Venujte pozornosť MAS vo fyzickom vývoji, na tvare prstov (prsty vo forme bubnových palice), zmeny v tvare hrudníka, charakteristické ochorenia spodných dýchacích ciest, hypopláziu lymfatických uzlín a mandlí.
Liečba.
Náhradná terapia: Intravenózne imunoglobulínové lieky sa zavádzajú každé 3-4 týždne pre život. Dávky imunoglobulínov sú vybrané tak, aby sa vytvorili v sére pacienta ich koncentrácie, prekrývajúc sa dolnú hranicu vekovej normy.
Diskutujte o možnosti génovej terapie - gén BTK.klonovaný, ale jeho hyperexpresia je spojená s malígnou transformáciou hematopoetickej tkaniny.
V prípade pretrvávajúcej neutropénie sa používajú rastové faktory. S výskytom príznakov autoimunitnej patológie je možné vymenovať lieky monoklonálnych protilátok (infliximab atď.).
Všeobecné variabilné imunitné zlyhanie
Bežné variabilné imunitné zlyhanie (Ovíin) je skupina syndrómov charakterizovaných defektom pre syntézu protilátok a bunkovú imunitu. Spoľahlivé diagnostické kritérium Avinu je významné zníženie obsahu imunoglobulínov dvoch alebo troch hlavných izotypov u ľudí oboch pohlaví v kombinácii s jedným z nasledujúcich znakov:
Debut choroby starších ako 2 roky;
Nedostatok izohemaglutinínov a / alebo nízkej odozvy očkovania;
Vylúčenie iných dôvodov pre AGHAMGLOBLOBULINEMIA.
U niektorých pacientov je príčinou vývoja Avinmu mutácia génov kódujúcich molekuly, ktoré sa podieľajú na procese dozrievania a prežitia B-buniek: BAFF-R (Receptor aktivujúceho B-bunky)BLIMP-1. (B-lymfocyt indukovaný proteín-1)a ICOS. (Indukovateľný kostimulátor).Existuje porušenie schopnosti B-lymfocytov diferencovať na plazmatické bunky, defekty protilátok sa vyvíjajú, t-lymfocytová dysfunkcia sa vyvíja, pozoruje sa zvýšená tendencia k infekčným ochoreniam. Syndróm sa môže prejaviť v ranom detstve, v adolescencii alebo mladých ľuďoch.
Laboratórne údaje.Hladiny IgG a IgA sa výrazne znižujú (približne 50% pacientov) a IgM (až do nepriebetného). Počet B-lymfocytov v krvi zodpovedá normu alebo zníženiu. Počet T-lymfocytov u väčšiny pacientov je normálny. U ťažkých pacientov je možné vývoj lymfopácie (menej ako 1500x103 bunky v 1 litri krvi). Počet NK buniek sa znižuje. Výroba špecifických protilátok v reakcii na imunizáciu je znížená alebo chýba. Výrazne porušujú proliferáciu lymfocytov a tvorba IL-2 pod pôsobením mitogénu a antigénov.
Klinický obraz.Prijímajte recidivujúce bakteriálne infekčné ochorenia s lokalizáciou prevažne v dýchacích cestách a neúplných dutín. V čase diagnózy respiračnej infekcie môže dýchacie trakty postupovať na bronchiektázu a rozliaty lézie ľahkého tkaniva. Je možné infekčné poškodenie tráviacich orgánov, ktoré sa prejavujú hnačkou, parníkom a malabsorpciou (a teda strata telesnej hmotnosti). Často identifikujú infekcie spôsobené Giardia Lamblia, Pneumocystis Cariniialebo rodinné vírusy Herpetiriridae.Pacienti AVIV sú náchylné na vývoj hnisavej artritídy spôsobenej mycoplazmom a uraplazmi. Prejavy enterovírusových infekcií môžu byť encefalomyelitída, polymické a dermatomy-podobné syndrómy, lézie kože a sliznice. Autoimunitnýchoroby sa vyskytujú tvrdo a môžu určiť prognózu ovinu. Niekedy prvé klinické prejavy AVIVE sú artritídou, nešpecifická ulcerózna kolitída a Crohnova choroba, skleníková cholangitída, malabsorpcia, SLE, nefritída, mositída, autoimunitné poškodenie pľúc vo forme lymfoidného pneumonitov, neutropénie,
trombocytopenic Purpura, Hemolytická anémia, zľavaná anémia, celková alopécia, retinálna vaskulitída, fotosenzibilizácia. U pacientov s ovinom sa frekvencia výrazne zvýši (v 15% prípadov), sarkoidos podobný granule a nezubandú lymfoproliferáciu. Liečba.
Antobakteriálna chemoterapia.
Náhradná terapia: Intravenózne imunoglobulínové lieky sa zavádzajú každé 3-4 týždne pre život.
V autoimunitných komplikáciách, imunosupresívna terapia (glukokortikoidy, azatioprín, cyklosporín A) a je možné predpísať monoklonálne prípravky protilátky (infliximab atď.).
Hyper-IgM Syndrómy
Hyper-IgM Syndrómy sú pomerne zriedkavé ochorenia charakterizované výrazným poklesom alebo úplnou neprítomnosťou IgG, IgA a normálnej alebo zvýšenej koncentrácie sérovej IgM. Je to spôsobené neschopnosťou B-lymfocytov na prepínanie tried imunoglobulínu a hypermutagenézy variabilných domén. K dnešnému dňu bolo identifikovaných 6 genetických defektov, čo viedlo k rozvoju hyper-igmsprom.
. Typ 1 (HIGM 1).Deficit Ligand X-Linked Ligand CD40 (70% syndrómov Hyper-IgM), čo vedie k neschopnosti T buniek, aby sa účinne interagovali s lymfocytmi.
. Typ 2 (HIGM 2).Autozomálne-recesívne, spojené s aktiváciou chýb indukovanej pomoci cytidínu deamindázu (gén AICDA, 12P13)- Enzým zapojený do spínacích tried imunoglobulínov a hypermutagenézy.
. Typ 3 (HIGM 3).Autozomálne recesívne, spojené s mutáciou génu molekuly CD40. Súčasne, B bunky sami nie sú schopní účinne interagovať s T-lymfocytom. Fenotypové prejavy sú podobné tým pre typ 1.
. Typ 4 (HIGM 4).Autozomálne recesívne; V niektorých prípadoch vznikajú mutácie de novo.Značilo sa s UNG - Enzýmom - enzýmom UNGIL-DNA-Glykosyláza, ktorá sa tiež zúčastňuje
v spínacích triedach imunoglobulínov, ale po akcii pomoci. V tomto prípade hypermutagenéza neovplyvní a syndróm prúdi s menšou závažnosťou.
. Typ 5 (HIGM 5).Porucha len v triedach spínania, hypermutagenéza neovplyvní. Príčinná mutácia ešte nebola zistená, ale samozrejme porucha enzýmu pôsobiaceho po
Pomoc.
. Typ 6 (HIGM-ED).X-hostované, spojené s výtlačnou ektodermálnou dyspláziou, spôsobenou nedostatkom NeMO (NF-KB modulátor), čo vedie k porušeniu prenosu signálu s CD40.
X-LUTED HYPER-IGM syndrómČastejšie. Vyvinutý, keď sa nachádza defekt CD40L kódovania génu (CD154, gén sa nachádza na XQ26-Q27.2)- ligand pre CD40. Insuficiencia expresie CD40L T-lymfocytov vedie k nemožnosti spínania tried imunoglobulín v B-lymfocytoch s IGM na iné izotypy, ako aj na porušenie tvorby B-buniek pamäte, repertoáru T-buniek a Th1 Celková odpoveď smerujúca proti intracelulárnym mikroorganizmom. Chlapci sú chorí
Laboratórne údaje.IgG, IgA, IgE nemožno určiť alebo detegovať vo veľmi malých množstvách. Úroveň IGM je normálna (v 50% prípadov) alebo zvýšená, často výrazne. Množstvo T- a B buniek je normálne; Znížená proliferatívna reakcia T buniek indukovaných antigénmi. IGM je polyklonálny, niekedy monoklonálny. Antentérapia IgM izotypu (anti-náhodné, antitromocyty, anti-náhodné, protilátky proti antigénom tkaniva hladkého svalstva). V lymfoidnej tkanine nie sú žiadne zárodočné centrá, ale existujú plazmatické bunky.
Klinický obraz.Prvé prejavy vznikajú v dojčenskej a ranom detstve. Charakteristický opakovaný infekciarôzne lokalizácia (primárne dýchacie cesty), vrátane oportunistickej (spôsobené Pneumocystis carinii).Tiež charakteristické pre vírusy (cytomegalovírus a adenovírusy), Criptococcus neoformans,mycoplazmy a mycobacteriums. Kryptosporidial infekcia môže spôsobiť akútnu a chronickú hnačku (vyvíjanie 50% pacientov) a sklerotizujúca cholangitídu. Anémia, neutropénia, ulcerácia ústnej sliznice, gingivitída, ulcerózna
včely pažeráka, rôzne črevné oddelenia, nešpecifická ulcerózna kolitída. Odhaliť predispozíciu voči K. autoimunitné poruchy(Séronegatívna artritída, glomerulonefritída atď.) A malígne neoplazmy (hlavne lymfoidné tkanivo, pečeň a žlčový trakt). Lymfadenopatia, hepato a splelamgaly sa môžu vyvinúť. Liečba
Pravidelná substitučná liečba intravenóznym imunoglobulínom.
Antobakteriálna chemoterapia. Na prevenciu a liečbu pneumóniových pneumónií sa používajú ko-trixazol [sulfametoxazol + trimetoprim] a pentamidín.
Aby sa zabránilo lézii pečene a žlčového traktu, sa má použiť iba varená alebo filtrovaná voda, pravidelne skúmať (ultrazvukové vyšetrenie, biopsia pečene podľa indikácií).
Pri liečbe neutropénia a ulcerácií ústnej dutiny sa používajú glukokortikoidy a prípravky granulocytov kolonystimilačný faktor.
Pri vývoji autoimunitných komplikácií, imunosupresívna terapia (glukokortikoidy, azatioprín, cyklosporín A), ako aj liečivá na báze monoklonálnych protilátok sú predpísané.
Optimálnym spôsobom liečby je transplantácia kostnej drene z darcov kompatibilných s HLA (úroveň prežitia je 68%, je lepšie vykonávať vo veku 8 rokov).
Kombinovaná imunodeficiencia s prevládajúcim defektom T-lymfocytom
Heavy kombinované imunitné zlyhanie
Tkin (scid - Ťažký kombinovaný imunitný nedostatok)- skupina syndrómov charakterizovaných znížením hladiny T-lymfocytov alebo ich úplnou absenciou a zhoršenou adaptívnou imunitou. . Retikulárna disgenéza, charakterizovaná porušením dozrievania lymfoidných a myeloidných predchodcov v počiatočných štádiách: neutropénia a T - B - NK -.
. X-Cutched tkin, vyvíjanie v dôsledku mutácie génu IL-2RG.[(CD132, celkom w.-CAKE receptor pre IL-2, IL-4, IL-7, IL-9, IL-15 a IL-21), XQ13.1-Q21.1,אּ ], ktorý vedie k blokáde receptorov a neschopnosť cieľových buniek, aby reagovali na pôsobenie zodpovedajúcich interleukínov (viac ako 50% všetkých prípadov tkin); T - B + NK -.
. Nedostatok tyrozínkinázy Janus3 [gén JAK3 (19P13.1),ρ ]; keď génové defekty, prenos aktivačného signálu zo všeobecného w.-Kake IL-2, IL-4, IL-7, IL-9, IL-15, IL-21 na jadro bunky, čo vedie k narušeniu diferenciácie T- a NK buniek; T - B + NK -.
. Insuficiencia proteintithosinfosfatázy (CD45, gén PTPRC, 1Q31-Q32);keď je defekt génu zvýšený inhibičnou aktivitou CSK kinázy na SRC proteintithiroidikinakinázou s porušením fosforylácie ITAM domén TCR a BCR; T - B + NK +.
. Úplný nedostatok enzýmov RAG1 a RAG2 aktivácia rekombinácie V (D) J-segmentov imunoglobulínov a TCR [génov Rag1a RAG2 (11P13),ρ ]; T - B - NK +.
. Syndróm je intenna (neúplný nedostatok enzýmov Rag1 a
RAG2) [gény Rag1a / alebo RAG2 (11P13-P12),r].Vďaka
nízka reziduálna aktivita týchto enzýmov sa stále vyvíja niekoľko t-lymfocytových klonov špecifických pre antigény epitelových tkanív kože a tráviaceho traktu, kde sa množia a produkujú veľké množstvá IL-4 a IL-5, čo spôsobuje hyperoeosinofylu a tvorba reziduálnych B-lymfocytov IgE (v neprítomnosti imunoglobulínov iných tried). Charakterizované erytrrodermia a pakidermy s alopéciou v pokožke hlavy a obočiam, vyčerpávajúce hnačku, ohrozujúce život infekčný syndróm; Hepatoslenomegaly a hyperplázové lymfatické uzliny.
. Tankin so zvýšenou citlivosťou na ionizujúce žiarenie. Defekt jadrového proteínu Artemis [gén Dclre1c, (10p),r],zahrnuté v komplexe enzýmov potrebných na opravu DNA (podieľa sa na zlúčenine dvojvláknových prestávok), s mutáciou génu, porušenia V (D) J-rekombinácie; T - B - NK +.
. Nedostatok IL-2 [gén IL-2, 4Q26-Q27].
Mutácia génu receptora receptora IL-2 (CD25) (10p15-P14);T - B + NK +.
Mutácie génu receptora receptora IL-7 (CD127) (5p13);T - B + NK +.
Nedostatok dechtu Transportér pre prezentáciu antigénu),potrebné antigénne peptidy v endoplazmatickom retikulóne génu IL-7 receptora (CD127) (5p13);T - B + NK +.
Mutácie génov CD3 reťazca (CD3y, CDA a CDε), čo vedie k zníženiu počtu zrelých T-lymfocytov, narušenia ich diferenciácie; T - B + NK +.
Nedostatočnosť proteindhirosinkinázy ZAP-70 [gén ZAP-70 (2Q12),r]. V mutácii génu, fosforylácia receptorov ITAM ζ-receptorov TCR a ITAM-obsahujúce NK bunkových bunkových receptorov trpí, selektívny nedostatok CD8 + T buniek sa vyvíja (obsah CD4 + T-lymfocytov je normálny, ale funkčný Poruchy sú vyjadrené vo forme nedostatku vzdelania týmito bunkami 2 a proliferácia).
Deficit formovania adenozínu [gén aDA (20Q12-Q13.11 , p), čo vedie k akumulácii v bunkách metabolitov (deoxyadenozidriphosfát a S-adenosylgomocysteín), inhibičná proliferácia T- a B-lymfocytov (možnosti pre neskorú debutu choroby); T - B - NK -.
Nedostatočnosť purrulucleozidefosforylázy [gén pnp (14Q11.2),p], čo vedie k akumulácii deoxyiguanosintriffosfátov v bunkách, ktoré inhibujú proliferáciu T-lymfocytov (sprievodné syndrómy - urrikamia a urikuria); T - B + NK -.
Laboratórne údaje.Nábor premennej, niekedy hlboký lymfopod; Lymfocyty nie sú schopné proliferovať v reakcii na špecifický antigén; Často sa často vyjadruje pokles hladiny imunoglobulínov v krvnom sére. Na rádiografii hrudníka nie je žiadny tieň Thymus.
Klinický obraz.Zvyčajne sa klinická diagnóza stáva jasnou v prvých 6 mesiacoch života, keď zmiznú materské IgG protilátky. V klinickom obrázku na poprednom mieste Ťažký infekčný syndrómlimfoidné tkanivo hypoplázia a vývojové oneskorenie. Infekčný syndróm sa vyznačuje perorálnou kandidóriou, chronickou hnačkou, pneumóniou, horúčkou,
septing bakteriálnej etiológie, vírusových infekcií. Infekcie infekcií patria do rôznych taxonomických skupín: baktérie, vírusy, huby, podmienečne patogénne mikroorganizmy (Pneumocystis carinii).Pneumónia je často spôsobená P. Carinii,hnačka - Rotavírus, Campylobacter, Giardia Lamblia.Často prejavuje vírusovú hepatitídu. Charakterizované rozvojom regionálnej alebo generalizovanej Bszhit po očkovaní.
Liečbastanovuje vymenovanie podpornej terapie vrátane parenterálnej výživy, zavedenia intravenózneho imunoglobulínu, účelu antibiotík, antifungálnych a antivírusových liekov. Jedným z hlavných spôsobov spracovania na dosiahnutie regenerácie - transplantácie kostnej drene, bez ktorej deti s tankinom zvyčajne umierajú v 1. ročníku života. Jednotlivé prípady sú opísané vtedy, keď dieťa žilo v obzvlášť dezinfikovaných podmienkach až 2-3 roky. Je dôležité rozpoznať tkin čo najskôr u novorodencov, ako je napríklad smrteľná, napríklad imunizácia živých vakcíny. Ihneď po nastavení diagnózy takýchto detí je potrebné vložiť do novelobiologických podmienok (sterilný box). V prípade pripojenia infekčných chorôb, intenzívnych anti-propatrial, antivírusovej a antifungálnej terapie, intravenózne imunoglobulínová substitučná terapia. Na prevenciu pneumatickej pneumónia je predpísaná ko-trimoxazol. V prípade vývoja Bvzhit je potrebné vykonávať dlhodobú intenzívnu intenzívnu anti-tuberkulózovú liečbu. Na transfúziu krvných zložiek by sa mali použiť len ožiarené a filtrované lieky. Existuje riziko post-transfúznej reakcie "transplantáciu proti hostiteľovi" v spojení s transplacentálnym prenosom materských lymfocytov.
Syndróm "nahé lymfocyty"
Takzvaná patológia, keď molekuly MHC-I alebo MNS-II nie sú exprimované v tele. Pri absencii expresie molekúl MHC-I sa zníži obsah CD8 + T-lymfocytov a neexistuje žiadna aktivita NK buniek; V neprítomnosti MNS-II sa zníži hladina CD4 + T-lymfocytov. Vyznačujú sa niektoré genetické defekty. Tieto chyby sú však lokalizované nie v génoch MHC, ale v niekoľkých rôznych faktoroch zodpovedných za reguláciu
výraz. Klinický obrazsyndróm "nahé" lymfocyty a liečbapodobné tým, ktorí so zvyškom tkin.
Djori syndróm
V syndróme Djori, alebo syndróm defektu defektov a štvrtého faryngeálnych vreckách [delécie v 22Q11vrátane génu TBX1 (22Q11,2),odhaliť hypopláziu alebo apláziu týmusu, padákovej žľazy hypoplasy, nedostatok srdca, nedostatok T-lymfocytov, variabilné množstvá B-lymfocytov.
Laboratórne údaje.Významný pokles množstva CD3 +, CD4 + a CD8 + T buniek a prudký pokles ich proliferatívnej aktivity indukovanej mitogénom a antigénom. Počet buniek B- a NK je normálny. Koncentrácia sérových imunoglobulínov Vo väčšine prípadov v normálnom rozsahu sú možné rôzne možnosti disbammaglobulinémie.
Klinický obraz.Komponent imunodeficiencie je reprezentovaná hypopláziou alebo skrinkou týmusu a opakujúce sa, vážne tečie infekčné choroby.Higoparathyroidizmus (hypokalcémiia a ako výsledok - totania, viditeľné 1-2 dni po narodení); Bezpečnosti obehového systému (pravé zvrátenie oblúka aorty, stenózy pravej komory, defektov v intervenickulárnych a interpretačných priečok, tetrad Fallo, Atresia alebo Helicoplasce svetlovej artérie); WPADIN NYABA; Anomálie kostry na tvár (zvýšená vzdialenosť medzi párovými orgánmi, redukovanými veľkosťami veľkostí, najmä nižšími, nízko plánovanými umývadlmi sluchu, krátke zásobníky). Vyjadrené abnormality štruktúry hrtanu, hltanxu, priedušnice, vnútorného ucha, pažeráka; Porušenie rozvoja obličiek, CNS a iných malformácií (polydaktilating, nedostatok nechtov, ANUS ATRESIA, ALÁLNA FISTULAS). Charakteristika je oneskorenie vývoja reči a psychomotorora. Berie na vedomie predispozíciu voči K. autoimunitné poruchy(Cytopénia, autoimunitná tyreoiditída) a malígne neoplazmy.
Liečba.. Antibakteriálna a antivírusová terapia. . Náhradná liečba intravenóznym imunoglobulínom. . Chirurgická liečba na účely korekcie malformácií. . V autoimunitných komplikáciách - imunosupresívna liečba. . Ak existujú endokrinopatie - korekcia vhodných porúch. . Transplantácia kostnej drene neeff
tivne. . Timurová transplantácia epiteliálneho tkaniva je odôvodnená. Korekcia funkcie parašitoidných okuliarov.
X-hákovaný lymfoproliferatívny syndróm
X- zachytený lymfoproliferačný syndróm je charakterizovaný porušením imunitnej reakcie na vírus Epstein-Barr [vďaka defektom génu SH2D1A.(SAP) v XQ25אּ], čo vedie k nekontrolovanej proliferácii B-lymfocytov transformovaných vírusom Epstein-Barr a infekciou vírusom s novými cieľovými bunkami.
Klinický obraz.4 najbežnejšie fenotypy sú opísané: Ťažká infekčná mononukleóza, malígne lymfoproliferatívne podmienky (lymfóm, leukémia, hlavne v bunke), anémii alebo šanca (vrátane v dôsledku hemopagocytového syndrómu indukovaného vírusom), disbammaglogulinémia. Infekcia vírusu Epstein-Barr je mechanizmus spúšťača (Launcher) na vytvorenie najťažších, rýchlo sa pohybujúcich a smrteľných ochorení: fulminantnej infekčnej mononukleózy (v 58% prípadov vedie k smrti), hemofagocytový syndróm (bez liečby v 100% prípadov vedie k fatálnemu výsledku). V 10% prípadov sa fenotyp prejavuje pred infekciou vírusom Epstein-Barr (vypúšťanie disbammaglobulinémie a lymfómu). Najčastejšie detekuje rôzne typy hypigamiglobulinémie. Imunodeficiencie vedie k rozvoju baktérií, plesní a vírusov infekčné choroby.Ochorenie môžete prevziať u chlapcov s charakteristickou rodinnou históriou a slúžili PCR pozitívnym testom na vírusoch Epstein-Barr. Na diagnostiku sa odporúča použiť kombináciu genetickej analýzy. SH2D1A.a odhady úrovne výrazu SAP.
Liečba
S cieľom prevencie sa odporúča používať antivírusové lieky - acyklovir, valacyclovar (ich skorý účel potláča replikáciu vírusu Epstein-Barr v Rothoglotku) a intravenóznom imunoglobulíne (s vysokým titerom protilátok proti vírusu Epstein-Barr).
V hypogammaglobulinémie sa intravenózny imunoglobulín používa mesačne v kombinácii s antibakteriálnou terapiou.
Vysoké dávky acykloviru a metylprednizolónu sú predpísané na liečenie fulminantnej infekčnej mononukleózy, vysoko kvalitnej liečby intravenóznym imunoglobulínom s vysokým titerom protilátok proti vírusu Epstein-Barr a IFNA.
S vývojom hemopagocytového syndrómu kombinuje vysoké dávky dexametazónu s semezidom ♠ (etoposid).
Pri liečbe malígnych ochorení sa používajú štandardné terapeutické protokoly.
Metóda radikálu - transplantácia kostnej drene z darcov kompatibilných s HLA.
Autoimunitný lymfoproliferatívny syndróm
Autoimunitný lymfoproliferatívny syndróm - skupina chorôb charakterizovaných benígnou lymfoproliferáciou, hyperimmunoglobulinémiou, autoimunitnými poruchami, zvýšeným obsahom CD3 + CD4 - CD8 - T-lymfocytov (dvojité negatívne) v periférnej krvi a defekt apoptózy [génu defekt Fas.(CD95) - TNFRRSF6 (10Q24.1),gól caspaza-10,Fas ligand - FASL (1Q23)].
Laboratórne údaje.Obsah CD3 + CD4 - CD8 - T-lymfocytov v periférnej krvi alebo v lymfoidných tkanivách je viac ako 1%. Hladina IgG, IgA a IgM môžu byť normálne, zvýšené alebo dokonca redukované. S vekom je hypergamamaglobulinémia nahradené nízkou koncentráciou sérových imunoglobulínov, až po Aghamglobulinmia. Odhaliť autoantibody na červené krvinky, krvných krviniek, krvných doštičiek, neutrofíl, hladké svaly, faktor VIII; Antinukleárne a anti-fosfolipidové autoprotilátky, ako aj reumatoidný faktor atď. Je charakterizovaný lymfocytózou.
Klinický obraz.Všetci pacienti majú zvýšenú pečeň, lymfatické uzliny (v prvých 5 rokoch života) a sleziny. Lymfoproliferácia nie je sprevádzaná zvýšením teploty a nočného potenia. Debutovať autoimunitné reakcienesmie sa zhodovať s lymfoproliferáciou a neskôr. S vekom sa zvyšuje závažnosť autoimunitných reakcií. Autoimunitné reakcie proti krvným bunkám (hemolytická anémia, trombocytopénia, neutropénia) sa vyvíjajú častejšie ako iné orgány. Riziko vzniku malígnych neoplazmov (T- a B-lymfóm, Berkittový lymfóm, atypický lymfóm, lymfogeumatóza atď.).
Liečba.. Chemoterapeutické činidlá (cyklosfamid, azatioprín, metotrexát, chlorambucil). . Glukokortikoidy. . Splenektómia s výrazným hypkupnizmom a hemocytopéniou. . V ťažkých prípadoch je možná transplantácia kostnej drene.
HYPERIMMUNOGLOOBULINEMIA Syndróm E.
Hyper-IgE syndróm je charakterizovaný významným zvýšením hladiny sérového IgE, opakovaných abscesov kože a podkožného vlákna stafylokokovej etiológie, pneumónia s tvorbou pneumatických, anomálií kostrovej štruktúry tváre ATD. Molekulárna genetická povaha hyper-IGN syndrómu ešte nebola stanovená. V niektorých prípadoch sa odhaľuje autozomalizovaný, v iných - autozomálne-recesívny dedičstvo. Predpokladá sa, že vady ovplyvňujú signalizačné molekuly cytokínových receptorov (v autozomálnej dominantnej forme tohto syndrómu, mutácie už boli odhalené Stat3)a prípadne spojené s porušením fungovania subpopulácie buniek TH17. Ďalší gén zodpovedný za tvorbu hyper-IGN syndrómu, lokalizovaný na chromozóme 4 (4Q).
Laboratórne údaje.Odhaliť rôzne imunologické poruchy: zvýšenie hladiny IgE v sére, zhoršená chemotaxia neutrofilov, defektov na tvorbu protilátok; Zníženie reakcie GZT na kandidínovú, difterésku a tetanickým anatoxínom; Oslabenie proliferatívnej aktivity T buniek v reakcii na Candida.a tetanus anatnoksín pri zachovaní odozvy na mitogens. Eozinofília v periférnej krvi a kvapalných kožných abscesoch. Počet T- a B buniek je normálny.
Klinický obraz.Ekzém strednodobého kurzu v ranom veku. Charakteristické vlastnosti tváre (širší most, široký nosný nos, asymetria tváre kostra, vyčnievajúce čelo, hlboko zasadené oči, vysoké podnebie). Anomálie pre rozvoj kostry, skoliózy, zvýšená mobilita kĺbov, tendenciu zlomenín kostí po menších zraneniach, porušenie zmeny zubov. Abscesy kože, podkožné vlákno a lymfatické uzliny vznikajú. Pneumónia sa vyvíja v staršom veku (najčastejšie patogény S. aureus.a H. \\ t
fluenzae),v 77% prípadov, pneumokele sa vytvorí, infekcia sa pripojí v dôsledku P. Aeruginosa.a A. Fumigatus.Pneumónia môže úniku bez zvýšenia teploty. Chronická kandidáza slizníc a nechtov sa vyvíja v 83% prípadov.
Liečba.. Dlhé (na účely prevencie - celoživotného) antibakteriálnej a antifungálnej terapie. . Na liečbu dermatitídy sa topické látky používajú v ťažkých prípadoch - nízke dávky cyklosporínu A .. Transplantácia kostnej drene je neúčinná.
Syndrómy rozkladu chromozómov
Pre syndrómy s chromozomálnou nestabilitou: aTTAKIATELEANGITASIA[Defekt génu DNA topoizomerázy Atm (11Q22),p] I. syndróm nijemegen[Defekt génu nibino Nbs1(8Q21)] - charakterizované zvýšenou frekvenciou malígnych nádorov, spontánnou chromozomálnou nestabilitou a chromozomálnymi poruchami. Obaja proteíny sa zúčastňujú na reparáciách medzier DNA a reguláciou bunkového cyklu. Normálne sa prestávky viažuce DNA vyskytujú v procese V (D) J rekombinácie imunoglobulínových génov a TCR, spínanie imunoglobulínových tried, počas zosieťovania a počas meyózy. Podobné procesy sa vyskytujú pri dozrievaní mozgových neurónov. DNA opravárenské defekty v ataxii-teleangection a Niimegenovom syndróme spôsobujú takéto klinické prejavy, ako je poškodená syntéza imunoglobulínu, funkcia genitálnych orgánov a nervového systému.
Ataxia Teleangitasia
Tento syndróm (frekvencia 1: 300 tisíc novorodencov) s veľmi heterogénnym fenotypom opísala francúzsky lekár D. Louis-bar. Symptómy ataxie možno nájsť u dieťaťa už vo veku 2-4 mesiacov. Ataxia je spôsobená progresívnou degeneráciou purkinierových buniek v cerebellum. Teleangectas na koži nosa, ucho-umývadlá, v spojivke sa objavujú o 3-6 rokov. Často na koži sú škvrny ako "káva s mliekom". Tymus hypoplasia, lymfatické uzliny, slezina, mandle sú charakteristické. Imunodeficiencia je znázornená znížením (často nerovnováha) IgA, IgE, IgG2, IgG4. Vyvíja sa 80% pacientov
vhodné infekčné klinické príznaky. Číslo a funkčná aktivita T buniek (najmä CD4 + T bunky) sa znižuje. Celkový počet T-lymfocytov u väčšiny pacientov je normálny. Extrémne vysoké (200-krát vyššie ako vo všeobecnej populácii) frekvencia neoplazmy (hlavne lymfóm a karcinómy), často vedúce k smrti o 10-12 rokov. Liečbasymptomatické.
Syndróm nijemegen
Syndróm Nijmegen (podľa názvu mesta v Holandsku, kde sa prvá choroba bola opísaná), ktorá sa prejavovala mikrocefalus, špecifický poškodený povrch tváre (skoseného čela, vyčnievajúce strednú časť tváre, dlhý nos, spodná čeľusť hypoplasia, mongolaid oko rezanie, Epicanti, veľké uši), oneskorenie fyzického rozvoja, prítomnosť hotelov "Káva s mliekom" na koži; Klianodectilia a syltons, dysgenéza vaječníkov a ďalších. Väčšina detí trpí recidivujúcou a chronickou baktériou infekčné chorobydýchacie cesty, lora orgánov a močový systém. V 50% prípadov sa vyvíjajú malígne neoplazmy, najmä v bunkových lymfómoch. Odhaľujú sa rôzne formy disbammaglobulinémie, zníženie CD4 + T-buniek.
Liečba.. Symptomatická liečba neurologických porúch. . Náhradná liečba intravenóznym imunoglobulínom. . Podľa indikácií sa používa antibakteriálne, antivírusové, antifungálne terapia. . Pri liečbe malígnych neoplazmov zohľadnila zvýšená citlivosť na žiarenie a chemoterapeutické výrobky.
Syndróm viscott-aldrich
Syndróm Viscott-Aldrich [Gene defekt WASP (XP11.23P11.22),אּ; tiež ρ a ʀ] gén Osa.(z Wiskott-Aldrich syndróm)vyjadrené v lymfocytoch, slezine a tymokytoch. Mutácie tohto génu sú spojené s abnormálnou expresiou v neutrofiloch a T-lymfocytoch (CD4 a CD8) CD43 molekulách (ligand pre ICAM-1, vykonáva anti-lepiacu funkciu).
Laboratórne údaje.Trombocytopénia (menej ako 10% normy) je spôsobená zvýšenou deštrukciou buniek.
Lístky ako u zdravých ľudí. Úroveň IgM v krvnom sére sa znižuje na normálnej úrovni IgG a zvýšenie obsahu IgA a IgE. Titre izohemagglutinínov sa znižujú, tvorba protilátok proti polysacharidovým antigénom pneumokokových, streptokokových, črevných tyčiniek, salmonely, ako aj antivírusových protilátok sa znižuje. V ranom veku je spravidla počet lymfocytov normálny, po 6 rokoch detekciu lymfoprodium (menej ako 1x109 / l), zníženie CD3 + a CD4 + T buniek na normálnej úrovni buniek V- a NK . Eozinofilia a vývoj Posthemorhagickej anémie sú možné. Je znížená proliferatívna reakcia T-buniek na mitogény a antigény, GZT je oslabený. V slezine nie sú zistené normálne štruktúry zárodočných centier a t-buniekových zón.
Klinický obraz.Choroba sa vyznačuje triadnými značkami: trombocytopénia, ekzém a opakované infekčné ochorenia.Hemoragický syndróm sa prejavuje skoro, už počas obdobia nováčikov (phetechial vyrážka, cheefarohematomes, krvácanie pupočníkových rany, črevného krvácania). Ekzém sa prejavuje od raného veku u 80% pacientov. Známky imunodeficiencie sa zvyšujú s vekom: bakteriálne infekčné ochorenia orgánov ENTRY, dýchacích ciest, tráviacich orgánov, kože; Bežná alebo zovšeobecná herpetická infekcia (Herpes simplexa Varicella Zoster),cytomegalovírus, ako aj plesň (kandidóza slizníc), menej často oportúnnych infekčných chorôb. U 70% pacientov identifikuje autoimunitné ochorenia (hemolytická anémia, neutropénia, artritída, kožná vaskulitída, nešpecifická ulcerózna kolitída, cerebrálna vaskulitída, glomerulonefritída, autoimunitná trombocytopénia). Pacienti nad 5 rokov zvýšili frekvenciu malígne neoplazmy(väčšinou tumor lemfoidný tkanivo).
Liečba.. Transplantácia alogénnych buniek kostnej drene alebo kmeňových buniek (úspech operácie dosahuje 90% pri použití štepu z histokompatibilného darcu a 50% - s haplozenténnou transplantáciou). . Náhradná liečba intravenóznym imunoglobulínom. . Preventívny účel antibakteriálnych, antifungálnych a antivírusových liečiv. . Na zníženie hemoragického syndrómu sa uskutočňuje splenektómia. . V autoimunitných komplikáciách je predpísaná imunosupresívna liečba.
Defekty fagocytózy
Chronické granulované ochorenie
Chronická choroba Granuner je charakterizovaná porušením funkčnej aktivity fagocytov (tvorba aktívnych foriem kyslíkových radikálov, intracelulárneho zabíjania a fragmentácie fagocyklovaných patogénov), trvalé bakteriálne a plesňové infekčné ochorenia a vývoj granulovaného zápalu. Chronické granulované ochorenia u osôb s rôznymi genetickými defektmi [v 65% prípadov - X-viazaná verzia ochorenia: gén gP91-PHOX (XP21.1),אּ; V 35% prípadov - autozomálne-recesívne: gén f47-PHOX (7Q11,23),ρ; gén p67-PHOX (1Q25),ρ; gén p22-PHOX (16Q24),p], čo vedie k porušeniu v systéme NADF-oxidázy. So smrťou krátkodobých (niekoľkých hodín) neutrofilov, neviazaných baktérií "úniku" do zamerania zápalu. MACROPHAGES - Dlhodobé bunky a ich predchodcovia (monocyty) migrujú na krb vo zvýšených množstvách (čo vedie k tvorbe granule)fagocytové mikroorganizmy, ale nie sú schopní ich zabiť.
Laboratórne údaje.Normálne hodnoty sérových imunoglobulínov a subpopulácie lymfocytov sú charakteristické. Tvorba peroxidátnych radikálov neutrofilmi sa odhaduje v testoch (chemiluminiscencia závislá od luminácie alebo obnovenie tetrazolium blue), je výrazne znížená alebo chýba. Proti pozadí infekčných chorôb, leukocytózy, neutrofilózy, zvýšenie ESR, anémie, hypergamaglobulinémie.
Klinický obraz.Choroba vo väčšine prípadov sa prejavuje v prvom roku života infekčný syndróm(Vnútorné infekcie a extracelulárne patogény) a tvorba granule. Najtypickejšie: Poškodenie pľúc (opakujúca sa pneumónia, lézia peších lymfatických uzlín, pľúcnych abscesov, hnisavých pleurisitov), \u200b\u200btráviaci trakt, kožné abscesy a lymfadentes. Najčastejšie patogény - kataláza-pozitívne mikroorganizmy: S. aureus, Aspergillus spp.,Črevné gram-negatívne baktérie (E. coli, Salmonella spp., Serratia Marcescens),menej pravdepodobné - Burkholderia Cepacia.a NoCardia Farcinica.Vývoj pečene a subatafragmálnych abscesov, osteomyelitídy, paragreeght abscesses, sepsis sú charakteristické.
Najviac závažná, život ohrozujúca infekčné komplikácie je aspergilóza, ktorá sa môže vyskytnúť vo forme difúznej lézie pľúc a iných orgánov (tukového tkaniva, mozgu, kostí, kĺbov, endokardu). U pacientov s chronickým granulovaným ochorením po očkovaní BCG často vyvíja infekciu súvisiacu s vakcínou, ktorá zahŕňa regionálne lymfatické uzliny. Mycobakteriálne poškodenie u pacientov s chronickým granulovaným ochorením môže mať ľahkú aj extrémnu lokalizáciu a zdĺhavý tok. Pre pacientov s chronickým granulovaným ochorením je lag vo fyzickom vývoji charakteristické.
Liečba.. Antimikrobiálna terapia: profylaktické konštantné použitie ko-trimuxazolu a antifungálnych liečiv (itrakonazol et al.); V prípade infekčných komplikácií sa uskutočňuje parenterálne kombinovaná antibakteriálna terapia (2-3 baktericídne antibiotiká penetračné intracelulárne) v kombinácii s antifungálnou terapiou. Pri vývoji aspergilózy sa zobrazí dlhodobé použitie amfotericínu v alebo kaspofung. Pri mykobakteriálnej infekcii sa používa kombinácia dlhodobej špecifickej terapie s liečivom anti-tuberkulózy so širokou škálou účinných antibiotík. . Chirurgická liečba je často sprevádzaná potlačením pooperačnej rany a tvorby nových ohniskách. Je možné prerušovaniu absorbujúceho abscesu pod kontrolou ultrazvuku. . Na liečbu ťažkých infekčných komplikácií v neefektívnosti antibakteriálnej terapie je možné použitie granulocytickej hmotnosti, vysoké dávky IFNU a G-CSF. . Transplantácia kostnej drene alebo transplantácia buniek káblov z kompatibilného súrodenca môže byť úspešná v ranom veku, keď je minimalizované riziko smrti proti infekčným komplikáciám a reakcii "transplantácia proti majiteľovi".
Adhézia defektu Leukocyte
K dnešnému dňu sú opísané 3 defekty leukocytov. Všetci majú
autosomal-recesívny typ dedičstva je charakterizovaný opakovaným a chronickým bakteriálnym a plesňovým infekčným ochoreniam. Typ I je charakterizovaný neprítomnosťou alebo znížením expresiou CD11 / CD18 na leukocytoch, prenikanie neutrofilov, leukocytózy (viac ako 25x10 9), neskôr depozícia zvyšku kábla a vývoj omphalitídy, chudobného hojenia rany, absencia tvorby hnisu pri peketraní cesty patogénu do tela.
Liečba.. Antibakteriálna terapia: infekčné epizódy a profylaktické. . V prípade ťažkého prúdu je predpísaná transplantácia kostnej drene z darcu kompatibilného HLA.
Vady systému komplementu
Choroby s deficitom komponentov doplnku
Prejavy génových defektov jednotlivých zložiek systému komplementu sú uvedené v tabuľke. 11-2.
Zdravý jsc.Choroby spôsobené deficitom komponentov komplementu sú zriedkavo detegované, pretože homozygotný stav autozomálnych alel je potrebný na ich prejav. Existuje jediné vylúčenie spojené s C1inH (inhibítor C1-esterázy): mutácia génu C1inh,inhibítor vedúci k deficitu v heterozygotnom stave sa prejavuje fenotypom, známym ako dedičný AO (pozri viac kapitoly 13, angio-pohybov).
Ochorenia imunitných komplexov.Insuficiencie C1-C4 sa prejavuje vývojom ochorení imunitných komplexov - systémové vaskulky a poškodenie obličiek, ktoré je zovšeobecnené nazývané syndróm systémového červeného lupusu (SC).
Pyiogénne infekcie.Nedostatok C3 (tiež faktory H a I) sú spojené so zvýšenou citlivosťou na pyrogénové infekcie. Deficit zložiek zapojených do alternatívnej cesty aktivácie komplementu, ako aj nedostatok zložiek C5-C8 spojených so zvýšenou citlivosťou na infekciu spôsobenú Neisseria spp.Nedostatok C9 je zvyčajne klinicky asymptoman.
Tabuľka 11-2.Klinické prejavy defektov jednotlivých zložiek systému komplementu
Lesten Deficit Lepenie Mannose
Lichen Deficitia spájajúci manóza (Iný názov: Mannanos LECTIN - MSL) je spôsobené defektom génu Mbl.(Rôzne bodové mutácie a delécie v géne Mbl.odhaliť zo 17% európskych ľudí pretekov). S vadami génu, aktivácia proteáz, rozdelenie zložiek komplementu C2 a C4 a aktivácia systému komplementu v dráhe lektínu je narušená. Klinickytáto patológia sa prejavuje infekčným syndrómom.
Laboratórne údaje.Analýza subpopulácií lymfocytov, leukocytov, ako aj imunoglobulínových izotypov nevykazuje významné odchýlky primerané klinickým symptómom. V sére nie je žiadna MSL.
Liečba.Táto choroba nie je klasická imunodeficiencia. V dôsledku toho je imunokorné imunotropné prostriedky kontraindikované. Rekombinantná MSL sa môže použiť ako farmakologický prípravok na etiopatogenetickú substitučnú liečbu pacientov s týmto dedičným defektom. V súčasnosti tento liek prechádza klinickými skúškami.
Prechodová hypogammaglobulinémia u detí
Prechodná hypogammaglobulinémia u detí je spojená s fyziologickým znakom fázovanej tvorby imunoglobulínového systému. Zrenie IgM a IgA protilátok je najviac "oneskorenia". U zdravých detí sa obsah materského IgG postupne znižuje a po pol roka zvyšuje vývoj vlastných IgG protilátok. V niektorých deťoch sa však zvýšenie úrovne imunoglobulínov oneskorí. Takéto deti môžu trpieť opakovanými bakteriálnymi infekčnými ochoreniami. V týchto prípadoch by sa nemalo uchýliť k infúziám liekov darcovských imunoglobulínov (zavedenie intravenózneho imunoglobulínu).
Selektívny imunoglobulínový nedostatok A
Selektívny imunoglobulín A (SD IGA - Selektívny nedostatok IGA)v dôsledku defektu génu tnfrsf13b.
alebo p). Deficit IgA v prítomnosti imunoglobulínov iných tried je najčastejšou imunodeficienciou zistenou v spoločnej populácii s frekvenciou 1: 500-1500 ľudí (u pacientov trpiacich alergiami, častejšie). IGA Selektívna insuficiencia, t.j. Prispievanie v nedostatku jednej zo podtried (30% prípadov) a kompletné (70% prípadov). Nedostatok podtriedy IGA2 vedie k výraznejšiemu klinickému obrazu ako deficit podtriedy IGA1. Kombinácie nedostatku IgA s inými porušeniami sú možné: s defektom IgG biosyntézy a anomálie T-lymfocytov. Prevažná väčšina osôb s selektívnym
deficit IgA je prakticky zdravý. Pre deti do 2 rokov je nedostatok IgA fyziologickým stavom.
Odhaliť zníženie koncentrácie séry IgA<5 мг/дл у детей старше 4 лет; IgG и IgM в норме, количество и соотношение субпопуляций лимфоцитов и их функциональная активность могут быть в норме.
Klinický obraz.S nedostatkom IgA, 3 skupiny patologických syndrómov sa môžu vyvinúť: infekčné, autoimunitné a alergické. Pacienti s nedostatkom IgA sú predisponované rekurentným infekčným ochoreniam horných dýchacích ciest a tráviacich orgánov. Najčastejšie a vážne tečie sú rôzne autoimunitné ochorenia (reumatoidná artritída, ankylozujúca spondylitída, Shegreen syndróm, vaskulitída s léziou mozgových ciev, autoimunitná tyreoiditída, SLE, glomerulonefritída, hemolytická anémia, diabetes typu I, vitiligo atď.). Frekvencia celiachood presiahne tak u detí s normálnou IGA 10-krát. Najčastejšie zistené alergické prejavy: neznášanlivosť k proteínu kravského mlieka, atopickej dermatitídy (ATD), bronchiálnej astmy.
Liečba.Prípady asymptomatického prietoku nevyžadujú žiadne špeciálne ošetrenie; V prítomnosti klinických prejavov infekčných, autoimunitných a alergických ochorení sa liečba vykonáva v súlade s normami.
Náhradná terapia imunoglobulínov darcov nie je znázornená v selektívnom, ani s úplnou nedostatkom IgA, pretože pravdepodobnosť tvorby anti-etehtiotických protilátok proti IGA a vývoj transfúznych komplikácií spôsobených nimi je vysoká.
Agamaglobulinémia s nedostatkom B-buniek
X-Cutched Aghamaglobulinémia (brutonova choroba)je to 90% všetkých prípadov AGHAMAGLOBULINEMIA. Chlapci sú chorí, synovia (אּ, ρ) nosičov chybného génu bTK (XQ21.3-Q22),b-lymfocytovo-špecifická proteindhyroskinkináza BTK (Brutonová tyrozínková kináza- Bruton Tyrozínkináza). V dôsledku vady, porušenie intracelulárnych signalizačných dráh, rekombinácie ťažkých imunoglobulínových reťazcov,
obnovenie trans B-buniek v B-lymfocytoch. U 10% pacientov s nedostatkom AGAMMAGLOBULINEmiA s nedostatkom v bunke, autosomonéza je zdedená. V súčasnosti je opísaných 6 genetických defektov, vrátane molekúl receptorov pred B-buniek, cytoplazmatického adaptérového proteínu B-buniek (BLNK) a génu Opakovanie bohatý na leucín (LRRC8).
Laboratórne údaje.Neexistujú žiadne periférne B-lymfocyty. V kostnej dreni sú pred-B bunky s μ-reťazcom v cytoplazme. Počet T-lymfocytov a funkčných testov na T-lymfocytoch môže byť normálne. IgM a IgA v krvi nemožno odhaliť; IgG môže byť prítomný, ale v malých množstvách (0,4-1,0 g / l). Žiadne protilátky proti krvným skupinám antigény a vakcínové antigény (tetanus, záškrtheria toxíny atď.). Neutropénia sa môže vyvinúť. Histologická štúdia lymfoidnej tkaniny: V lymfoidných folikulách nie sú žiadne zárodočné (zárodočné) centrá a plazmatické bunky.
Klinický obraz.Ak je rodinná história neznáma, diagnóza sa v priemere zrejmé ako 3,5 roka. Choroba sa vyznačuje lymfoidným tkanivom hypoplasia, silne tečúcou hnisavými infekciami, infekčnými ochoreniami horných (sinusites, otitis) a nižšia (bronchitída, pneumónia) dýchacích ciest; Gastroenteritída, pyodermia, septická artritída (bakteriálna alebo chlamydious), septikémia, meningitída, encefalitída, osteomyelitída. Ako kauzatívne činidlá respiračných ochorení, najčastejšie vykonávané Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus,hnačka črevnej baktérie alebo giardia Giardia Lamblia.Pacienti s Aghamaglobulinémiou sú tiež citlivé na infekčné ochorenia spôsobené mykoplazmom a uraplazmi, ktoré spôsobujú vývoj chronických pneumónia, hnisavých artritídy, cystitu a subkutánneho abscesu. Z vírusov typických neurotropných vírusov ECHO-19 a kokes, čo spôsobuje ťažkú \u200b\u200bostrú a chronickú encefalitídu a encefalomyelitídu. Prejavy enterovírusových infekcií môžu byť syndróm ako dermatomy, ataxia, bolesti hlavy, porušovanie správania. U pacientov s deťmi v imunizácii, živý polyovakcinát, spravidla, dlhodobo uvoľňovanie vírusu POLOŽE sa deteguje prostredníctvom slizníc a s obnovenou a zvyšujúcou sa virulenciou (t.j. v detskom kolektore
sme skutočným nebezpečenstvom infekcie zdravých detí s POLOHA, v dôsledku kontaktu s očkovaným imunodeficientným dieťaťom). Autoimunitné poruchy počas agamaglobulinémie môžu byť reprezentované reumatoidnou artritídou, syndrómou podobným sklerermovým, scrugarl, nešpecifický ulcerózna kolitída, diabetes typu I (kvôli prevahu thl imunitnej reakcie).
Fyzickú kontrolu.Venujte pozornosť MAS vo fyzickom vývoji, na tvare prstov (prsty vo forme bubnových palice), zmeny v tvare hrudníka, charakteristické ochorenia spodných dýchacích ciest, hypopláziu lymfatických uzlín a mandlí.
Liečba.
Náhradná terapia: Intravenózne imunoglobulínové lieky sa zavádzajú každé 3-4 týždne pre život. Dávky imunoglobulínov sú vybrané tak, aby sa vytvorili v sére pacienta ich koncentrácie, prekrývajúc sa dolnú hranicu vekovej normy.
Diskutujte o možnosti génovej terapie - gén BTK.klonovaný, ale jeho hyperexpresia je spojená s malígnou transformáciou hematopoetickej tkaniny.
V prípade pretrvávajúcej neutropénie sa používajú rastové faktory. S výskytom príznakov autoimunitnej patológie je možné vymenovať lieky monoklonálnych protilátok (infliximab atď.).
Všeobecné variabilné imunitné zlyhanie
Bežné variabilné imunitné zlyhanie (Ovíin) je skupina syndrómov charakterizovaných defektom pre syntézu protilátok a bunkovú imunitu. Spoľahlivé diagnostické kritérium Avinu je významné zníženie obsahu imunoglobulínov dvoch alebo troch hlavných izotypov u ľudí oboch pohlaví v kombinácii s jedným z nasledujúcich znakov:
Debut choroby starších ako 2 roky;
Nedostatok izohemaglutinínov a / alebo nízkej odozvy očkovania;
Vylúčenie iných dôvodov pre AGHAMGLOBLOBULINEMIA.
U niektorých pacientov je príčinou vývoja Avinmu mutácia génov kódujúcich molekuly, ktoré sa podieľajú na procese dozrievania a prežitia B-buniek: BAFF-R (Receptor aktivujúceho B-bunky)BLIMP-1. (B-lymfocyt indukovaný proteín-1)a ICOS. (Indukovateľný kostimulátor).Existuje porušenie schopnosti B-lymfocytov diferencovať na plazmatické bunky, defekty protilátok sa vyvíjajú, t-lymfocytová dysfunkcia sa vyvíja, pozoruje sa zvýšená tendencia k infekčným ochoreniam. Syndróm sa môže prejaviť v ranom detstve, v adolescencii alebo mladých ľuďoch.
Laboratórne údaje.Hladiny IgG a IgA sa výrazne znižujú (približne 50% pacientov) a IgM (až do nepriebetného). Počet B-lymfocytov v krvi zodpovedá normu alebo zníženiu. Počet T-lymfocytov u väčšiny pacientov je normálny. U ťažkých pacientov je možné vyvinúť lymfopéniu (menej ako 1500x103 bunky v 1 litri krvi). Počet NK buniek sa znižuje. Výroba špecifických protilátok v reakcii na imunizáciu je znížená alebo chýba. Výrazne porušujú proliferáciu lymfocytov a tvorba IL-2 pod pôsobením mitogénu a antigénov.
Klinický obraz.Prijímajte recidivujúce bakteriálne infekčné ochorenia s lokalizáciou prevažne v dýchacích cestách a neúplných dutín. V čase diagnózy respiračnej infekcie môže dýchacie trakty postupovať na bronchiektázu a rozliaty lézie ľahkého tkaniva. Je možné infekčné poškodenie tráviacich orgánov, ktoré sa prejavujú hnačkou, parníkom a malabsorpciou (a teda strata telesnej hmotnosti). Často identifikujú infekcie spôsobené Giardia Lamblia, Pneumocystis Cariniialebo rodinné vírusy Herpetiriridae.Pacienti AVIV sú náchylné na vývoj hnisavej artritídy spôsobenej mycoplazmom a uraplazmi. Prejavy enterovírusových infekcií môžu byť encefalomyelitída, polymické a dermatomy-podobné syndrómy, lézie kože a sliznice. Autoimunitnýchoroby sa vyskytujú tvrdo a môžu určiť prognózu ovinu. Niekedy prvé klinické prejavy AVIVE sú artritídou, nešpecifická ulcerózna kolitída a Crohnova choroba, skleníková cholangitída, malabsorpcia, SLE, nefritída, mositída, autoimunitné poškodenie pľúc vo forme lymfoidného pneumonitov, neutropénie,
trombocytopenic Purpura, Hemolytická anémia, zľavaná anémia, celková alopécia, retinálna vaskulitída, fotosenzibilizácia. U pacientov má ovína výrazne zvýšená frekvencia malígne neoplazmy(V 15% prípadov), sarkoidos, ako je granula a nezubandová lymfoproliferácia. Liečba.
Antobakteriálna chemoterapia.
Náhradná terapia: Intravenózne imunoglobulínové lieky sa zavádzajú každé 3-4 týždne pre život.
V autoimunitných komplikáciách, imunosupresívna terapia (glukokortikoidy, azatioprín, cyklosporín A) a je možné predpísať monoklonálne prípravky protilátky (infliximab atď.).
Hyper-IgM Syndrómy
Hyper-IgM Syndrómy sú pomerne zriedkavé ochorenia charakterizované výrazným poklesom alebo úplnou neprítomnosťou IgG, IgA a normálnej alebo zvýšenej koncentrácie sérovej IgM. Je to spôsobené neschopnosťou B-lymfocytov na prepínanie tried imunoglobulínu a hypermutagenézy variabilných domén. K dnešnému dňu bolo identifikovaných 6 genetických defektov, čo viedlo k rozvoju hyper-igmsprom.
. Typ 1 (HIGM 1).Deficit Ligand X-Linked Ligand CD40 (70% syndrómov Hyper-IgM), čo vedie k neschopnosti T buniek, aby sa účinne interagovali s lymfocytmi.
. Typ 2 (HIGM 2).Autozomálne-recesívne, spojené s aktiváciou chýb indukovanej pomoci cytidínu deamindázu (gén AICDA, 12P13)- Enzým zapojený do spínacích tried imunoglobulínov a hypermutagenézy.
. Typ 3 (HIGM 3).Autozomálne recesívne, spojené s mutáciou génu molekuly CD40. Súčasne, B bunky sami nie sú schopní účinne interagovať s T-lymfocytom. Fenotypové prejavy sú podobné tým pre typ 1.
. Typ 4 (HIGM 4).Autozomálne recesívne; V niektorých prípadoch vznikajú mutácie de novo.Značilo sa s UNG - Enzýmom - enzýmom UNGIL-DNA-Glykosyláza, ktorá sa tiež zúčastňuje
v spínacích triedach imunoglobulínov, ale po akcii pomoci. V tomto prípade hypermutagenéza neovplyvní a syndróm prúdi s menšou závažnosťou.
. Typ 5 (HIGM 5).Porucha len v triedach spínania, hypermutagenéza neovplyvní. Príčinná mutácia ešte nebola zistená, ale samozrejme porucha enzýmu pôsobiaceho po
. Typ 6 (HIGM-ED).X-hostované, spojené s výtlačnou ektodermálnou dyspláziou, spôsobenou nedostatkom NeMO (NF-KB modulátor), čo vedie k porušeniu prenosu signálu s CD40.
X-LUTED HYPER-IGM syndrómČastejšie. Vyvinutý, keď sa nachádza defekt CD40L kódovania génu (CD154, gén sa nachádza na XQ26-Q27.2)- ligand pre CD40. Insuficiencia expresie CD40L T-lymfocytov vedie k nemožnosti spínania tried imunoglobulín v B-lymfocytoch s IGM na iné izotypy, ako aj na porušenie tvorby B-buniek pamäte, repertoáru T-buniek a Th1 Celková odpoveď smerujúca proti intracelulárnym mikroorganizmom. Chlapci sú chorí
Laboratórne údaje.IgG, IgA, IgE nemožno určiť alebo detegovať vo veľmi malých množstvách. Úroveň IGM je normálna (v 50% prípadov) alebo zvýšená, často výrazne. Množstvo T- a B buniek je normálne; Znížená proliferatívna reakcia T buniek indukovaných antigénmi. IGM je polyklonálny, niekedy monoklonálny. Antentérapia IgM izotypu (anti-náhodné, antitromocyty, anti-náhodné, protilátky proti antigénom tkaniva hladkého svalstva). V lymfoidnej tkanine nie sú žiadne zárodočné centrá, ale existujú plazmatické bunky.
Klinický obraz.Prvé prejavy vznikajú v dojčenskej a ranom detstve. Charakteristický opakovaný infekciarôzne lokalizácia (primárne dýchacie cesty), vrátane oportunistickej (spôsobené Pneumocystis carinii).Tiež charakteristické pre vírusy (cytomegalovírus a adenovírusy), Criptococcus neoformans,mycoplazmy a mycobacteriums. Kryptosporidial infekcia môže spôsobiť akútnu a chronickú hnačku (vyvíjanie 50% pacientov) a sklerotizujúca cholangitídu. Anémia, neutropénia, ulcerácia ústnej sliznice, gingivitída, ulcerózna
včely pažeráka, rôzne črevné oddelenia, nešpecifická ulcerózna kolitída. Odhaliť predispozíciu voči K. autoimunitné poruchy(Séronegatívna artritída, glomerulonefritída atď.) A malígne neoplazmy (hlavne lymfoidné tkanivo, pečeň a žlčový trakt). Lymfadenopatia, hepato a splelamgaly sa môžu vyvinúť. Liečba
Pravidelná substitučná liečba intravenóznym imunoglobulínom.
Antobakteriálna chemoterapia. Na prevenciu a liečbu pneumóniových pneumónií sa používajú ko-trixazol [sulfametoxazol + trimetoprim] a pentamidín.
Aby sa zabránilo lézii pečene a žlčového traktu, sa má použiť iba varená alebo filtrovaná voda, pravidelne skúmať (ultrazvukové vyšetrenie, biopsia pečene podľa indikácií).
Pri liečbe neutropénia a ulcerácií ústnej dutiny sa používajú glukokortikoidy a prípravky granulocytov kolonystimilačný faktor.
Pri vývoji autoimunitných komplikácií, imunosupresívna terapia (glukokortikoidy, azatioprín, cyklosporín A), ako aj liečivá na báze monoklonálnych protilátok sú predpísané.
Optimálnym spôsobom liečby je transplantácia kostnej drene z darcov kompatibilných s HLA (úroveň prežitia je 68%, je lepšie vykonávať vo veku 8 rokov).
Toto je najčastejšia primárna imunodefickosť. Vyskytuje sa s frekvenciou 1/300 - 1/700. Najprv bol ...
Opísal J. Heremans v roku 1960.
Vo väčšine prípadov, jasný obraz o dedičstve ochorenia nie je pozorovaný, vo viacerých zriedkavých prípadoch straty rodiny IgA, autozomálne-recesívne dedičstvo nastane častejšie ako dominantné. Deficit IgA je niekedy spojený s chromozómovými anomáliami 18.
IgA sa líši od iných imunoglobulínov v obsahu sacharidov a kyseliny sialovej a schopnosť vytvárať diméry, triméry a dokonca aj tetraméry. Rozlišovať 2 možnosti pre molekuly IgA:
- Sérum IgA - vždy monomér;
- Secretor IgA - tvorí di-, tri a tetraméry, pred tým, než sa stanú neoddeliteľnou súčasťou tajomstiev (sliny, slza, mlieko, tajomstvo tráviacich, dýchacích ciest a močových ciest).
Špeciálny proteínový reťazec sa zúčastňuje na vzdelávaní sekrečnej IGA - J-reťazec. V dôsledku toho je to celý proteínový systém. Odtiaľ - formuláre patológie IGA:
- Celkové zlyhanie IGA je spojené s anomáliami syntézy monomérov IgA. Výsledkom je, že obsah a sérum a sekrečné IGA sú znížené. Miestna a celková ochrana je narušená;
- vada na vytvorenie sekrektových molekúl - SIGA. Dôvodom môže byť absencia J-reťazca, ktorá vedie k porušeniu miestnej imunity;
- Preferenčné porušenie syntézy séra IgA, keď sú pridelené iba sekrečné IgA formy a monoméry sérových IgA sú prakticky vylučované.
Klinické príznaky závisia od igA Deficience Times A môžu sa prejaviť vo forme patológie jednotlivých orgánov a systémov. Najčastejšie sú infekčné poškodenie slizníc.
Z hlavy gastrointestinálneho traktu je chronická gastritída, ulcerózna a hemoragická kolitída, ileit, stomatitída. Z boku dýchacích ciest - rinitída, sinusites, bronchitída, rýchlo dostávajú chronický charakter. Často sú chronické bronchopneumónia s výsledkom v bronchiectáze a pľúcnych abscesoch. Spojenie zníženého obsahu sekrečnej IgA so zvýšenou morbiditou atopických alergií bol tiež detegovaný a s vývojom autoimunitných ochorení (systémový červený lupus, reumatoidná artritída, autoimunitná tyreoiditída).
V osôb so selektívnym deficitom prevažneho sérového monoméru IgA sa vyskytujú najčastejšie žiadne klinické prejavy imunodeficiencie.
Laboratórny výskum:ukázalo sa, že celkový počet T- a B-lymfocytov v periférnej krvi u týchto pacientov je v normálnom rozsahu. Sérum a / alebo sekrečné IgA hladiny sú ostro znížené, zatiaľ čo obsah IGM a IgG je v normálnom rozsahu. U jednotlivých pacientov (s prejavmi atopických alergií) môže byť hladina IgE zvýšená.
Liečba: symptomatické. Je potrebné byť opatrní pri použití substitučnej terapie imunoglobulínom, pretože keď opakované podávanie existuje riziko anafylaktického šoku kvôli možnosti zvýšených anti-imunoglobulínových produktov IgG a tried IgM.
Prognóza: Kurz choroby je hlavne benígny.
Diagnostické kritérium - Znížené u pacientov počas 4 rokov hladín IgA sérových hodnôt menej ako 0,07 g / l s normálnym obsahom IgG a IgM a vylúčenie iných príčin hypogamihlobulínmií. Diagnosticky významné:
Izolované redukcia hladín sérových IgA (menej ako 0,05 g / l) na normálnej úrovni iných izotypov imunoglobulínu u detí starších ako 1 rok, nie IGAL a IGA2. Úrovniami IGM a IgG normálne. Niektorí pacienti však identifikujú nedostatok IgG2;
- ak je hladina IgA v rozsahu 0,05 g / l do 0,2 g / l, potom je diagnostikovaná čiastočná nedostatočnosť IgA; normálny počet T-lymfocytov a ich podtriedy;
- zvyčajne normálny počet in-lymfocytov (CD19 CD20);
- Normálna NK CELL (CD16 CD56).
U pacientov s nedostatkom IgANajmä v neprítomnosti sekrečnej IGA je potrebné preskúmať úroveň podtriedy IgA. U niektorých pacientov môže selektívna insuficiencia IgA napredovať s rozvojom v budúcnosti AVIV. Je potrebné dlhé pravidelné monitorovanie obsahu imunoglobulínov (vrátane asymptomatických pacientov).
Stanovenie autorantilnej (antinukleárnej, antidididy atď.).
S jedlom neznášanlivosť Alebo malabsorpcia potrebuje alergiu a definovanie protilátok proti mliečnym a anti-nežiaducim IgG protilátkam.
Liečba. Pacienti s asymptomatickým selektívnym nedostatkom konštantnej liečby IgA sa nevyžaduje. Pacienti s prejavmi infekčných ochorení sú predpísané profylaktickým cieľom antibiotík. Intenzívne antibakteriálne spracovanie sa uskutočňuje u všetkých pacientov počas výskytu infekčného ochorenia. Pacienti nie sú kontraindikovaná rutinná imunizácia. Náhradná imunoglobulínová terapia je kontraindikovaná, keď pacient detekuje autoantibódy proti IGA. Je potrebné vziať do úvahy skutočnosť, že volebný nedostatok IgA označuje neorozívne primárne imunitné chyby. Terapeutické opatrenia sa znižujú na symptomatickú liečbu infekčných, alergických a autoimunitných ochorení. Imunotropné liečivá sú predpísané hlavne v dôsledku prejavu zvýšeného incidencie.
Prognóza. U pacientov so selektívnym nedostatkom IgA, prognóza závisí od prítomnosti súbežného defektu špecifických protilátok, alergií alebo autoimunitných ochorení. Asymptomatický priebeh ochorenia môže byť často narušený v dôsledku pôsobenia externých ovplyvňujúcich faktorov, napríklad v stresovej situácii, v imunosupresii, chemoterapii atď.

