Debe diferenciarse del síndrome real durante la hipervitaminosis, en la que, como resultado de la insuficiencia enzimática del canal de riñón, se están desarrollando trastornos metabólicos graves en el cuerpo del niño. V. V. Shitskova y Sovat. (1971) Sobre la base de sus propias observaciones, la siguiente tabla de diagnóstico diferencial de estas enfermedades conduce (Tabla 7) con algunas de nuestras adiciones.
| Indicadores | Hipervitaminosis D. | Síndrome de Tony-Debe Fanconi |
| Frecuencia | Sin importar | Raramente |
| Patogénesis | Violación de los procesos metabólicos, principalmente calcio, debido a la sobredosis de vitamina D | Enzimopatía. Tubulopatía congénita. Phosphorus reabsorción violación, glucosa y aminoazot. |
| Cuadro clinico | Sequedad y palidez Piel, sed, vómitos, estreñimiento, hipótrofia, hipertensión, aumento del hígado | Suhness y Palores Piel, Anorexia, Sed, Vómitos, Estreñimiento, Poliuria, Hipotrofia, Aumento del hígado. La hipertensión no es. Hipotensión muscular |
| Análisis de sangre bioquímica | Hipercalcemia en el período agudo. Fósforo bajó. Azúcar y proteína normalmente. La fosfatasa alcalina no se cambia | El calcio es normal o reducido. El fósforo se reduce bruscamente. El azúcar y la proteína se reducen, la actividad de la fosfatasa alcalina ha aumentado considerablemente. Acidosis metabólica |
| Orina | La reacción Sulkovic es positiva. Proteinuria, microhematuria, leucocituria. El azúcar aminoazot es más a menudo normal. | La reacción Sulkovich es negativa. Proteinuria, Fosfaturia, Glucosuria, AminoAciduria |
| Radiografía de huesos tubulares. | Expansión y sellado de zonas preliminares de observación. | Osteoporosis de huesos tubulares, zonas ocasionales de pobres. |
Síndrome de Fanconi. - Diabetes amina. Trastorno de cambio de cistina, acompañado de glicosuria, aminoaciuría, fosfatina, mayor alineación, pérdida alcalina. Debido al defecto hereditario de los túbulos renales, causando la imposibilidad de la absorción inversa de glucosa, aminoácidos y fosfato. El intercambio de cistina está frustrado, que se pospone en forma de cristales en el sistema endotelial reticular, córnea, túbulos renales y otros tejidos. Esto conduce a la insuficiencia progresiva de varias funciones de los túbulos renales.
El síndrome de Fanconi de la cistinuria debe distinguirse, a la que la cistina de los tejidos no se pospone. En el síndrome, la capacidad funcional del aparato glomerular sigue siendo normal. La concentración de calcio en suero es normal, el nivel de fósforo inorgánico disminuye considerablemente. En la radiografía, se observan osteomalacia y agotamiento difuso de los álcali los álcali.
El síndrome (enfermedad) de Tony - Debe - Fanconi es una de las enfermedades similares a los raquetes.
Historia
La síndrome de riñón fue identificado por el pediatra suizo Fanconi entre otros investigadores descritos anteriormente partes separadas Enfermedades. En 1931, describió al niño con enjuague y raquitismo glucosuria y albuminuria, 2 años después, de Tony agregó a cuadro clinico Los hipofosfatems, y pronto la debbre describió la aminoaciduria.
Etiopatogénesis
El tipo de herencia es un reciclaje autosómico, también se resalta una forma autosómica dominante con una localización de genes en el cromosoma 15q15.3. La expresividad del gen mutante en estado homocigoto varía significativamente. Hay casos esporádicos debido a la mutación fresca. Se cree que la enfermedad se basa en defectos determinados genéticamente de la fosforilación enzimática en los canales renales (tubulopatía combinada), la escasez de enzimas de los 2º y 3º complejos de la cadena respiratoria: succinatdeehidrogenasa y citocromoxidasa. Varios autores consideran esta enfermedad a la categoría de enfermedades mitocondriales.
Las causas descritas anteriormente conducen a la violación de los procesos de la fuente de alimentación del transporte de fosfatos, la glucosa y los aminoácidos en los túbulos renales y su mayor excreción con la orina, así como al trastorno de los mecanismos para mantener el equilibrio de la base ácida. El desarrollo de acidosis metabólica y desventaja de los compuestos de fosfato contribuye al deterioro de la formación tejido óseo De acuerdo con el tipo de osteomálisis y cambios similares a los riquetas del esqueleto.
Cuadro clinico
Los primeros signos aparecen en la segunda mitad del primer año de vida, el complejo de síntomas desplegado está formado por el segundo año de vida. A veces la manifestación es de 6 a 7 años. Signos iniciales - Sed, poliuria, vómitos, a veces un largo subfebelitet. En el segundo año detectan el atraso. desarrollo fisico y las deformaciones óseas de las extremidades inferiores (Valgusnia o Vius), pecho, antebrazo I. huesos, reducción del tono muscular. El Determine radiológicamente la osteoporosis sistémica de diversos grados de gravedad, el adelgazamiento de la capa cortical de los huesos tubulares, rompiendo las zonas de crecimiento, el retraso de la tasa de crecimiento del tejido óseo de la era biológica del niño. Los huesos se vuelven frágiles.
Dependiendo de la severidad manifestaciones clínicas y los trastornos metabólicos asignan dos variantes clínicas y bioquímicas de la enfermedad. Primero Se caracteriza por un retraso significativo en el desarrollo físico, un curso serio de la enfermedad con deformaciones óseas severas y, a menudo, fracturas de huesos, hipocalcemia de corte (1.6-1.8 mmol \\ l), disminución de la absorción de calcio en el intestino. Para segundo La variante señala un retraso moderado en el desarrollo físico, un flujo de luz con deformaciones de huesos menores, normocalcemia y asimilación normal de calcio en el intestino.
Violaciones bioquímicas
- reducción de los niveles de calcio en la sangre;
- reduciendo el nivel de fósforo en la sangre;
- mejorar el nivel de fosfatasa alcalina;
- el desarrollo de la acidosis metabólica (pH \u003d 7.35-7.25; ve \u003d -10 ... -12 mmol \\ l) debido al defecto de la reabsorción de bicarbonatos en los túbulos proximales;
- excreción normal de calcio con orina;
- el aumento del aclaramiento de fosfato de orina, la absorción de fosfato en el intestino no sufre;
- desarrollo de glucosuria (20-30 g \\ l y superior);
- el desarrollo de hiperámicaisduria generalizada;
- violación de las funciones de amonioacidogénesis: una disminución en la acidez de la titulación, un aumento en el pH de la orina es mayor que 6.0;
- desarrollo de la hipopotasemia.
Diagnóstico diferencial
La enfermedad de Tony-Debra-Fanconi debe diferenciarse con síndrome secundario, detectado en otras enfermedades hereditarias y adquiridas (
Síndrome de Fanconi (nombre completo - de Tony-Debre Fanconi) - Patología congénita, que se expresa en una disfunción grave de túbulos renales proximales, a saber, una violación de la absorción secundaria (succión en la sangre) filtrada por las sustancias renales que conduce a la glucosuria ( aumento del azúcar En la orina), fosfaturia (violación de fósforo y metabolismo de calcio), aminoacidurio (aumento de la eliminación de aminoácidos con orina) y reduciendo la concentración de hidrocarburos que gobiernan la acidez de la sangre.
Síndrome de Tony-Debe Fanconi
Síndrome de Fanconi por lo enfermedad rara, en su mayoría ocurriendo en niños, y estadísticas médicas Su frecuencia corresponde a 1 paciente al bebé para 350 mil recién nacidos de ambos sexos.
En los adultos, las personas son extremadamente raras, desarrollándose contra el fondo de las patologías adquiridas. Código de patología según ICD-10: E72.O.
Las razones
La naturaleza y las causas del vicio genético de síndrome de Fanconi hoy no están suficientemente estudiadas.
Se supone que la patología se basa en o vicios de proteínas de transporte de túbulos renales o mutación del gen, distorsionando la función de las enzimas que regulan la absorción inversa de glucosa, aminoácidos y fósforo.
Hay datos de la investigación sobre defectos de puntos de mitocondria, lo que lleva al funcionamiento incorrecto de los canales de riñón.
La enfermedad también está asociada con la intolerancia a la fructosa, envenenamiento crónico Toxinas (metales pesados, iphosfamida, aminoglucósidos), deficiencia de vitamina D, amiloidosis, insuficiencia de una serie de enzimas celulares (piruvaturboxilasa, fosfoenolpyuruaturbojesas, y otros), tirosinemia, leukeodor, galactosemia, cistinosis, glicogenesis.
Según otros especialistas, el síndrome de Fanconi se puede aislar la patología, es decir, una de las formas pesadas de las patologías similares a los raquitis que tienen herencia.
Los estudios confirman que en el síndrome de Fanconi, el intercambio de energía celular que involucran el ATP (trifosfato de adenosina) y el transporte intercelular en los túbulos del elemento de riñón principal: se perturban la nefrona.
Debido a la falta de funciones de enzimas, glucosa, fosfatos, se están produciendo aminoácidos, y los canales de riñón están experimentando déficit de energía. Al mismo tiempo, las sustancias importantes van con la orina, lo que lleva a cambios distróficos en el tejido óseo: raquitis.
Dado que el medicamento aún no ha llegado a una conclusión inequívoca sobre las causas del síndrome de Fauxy, esta condición se denota por otros términos: "diabetes de glucoposfamina", "síndrome de ventilador renal idiopático", resistente a D Rahit "," Nanismo renal con D- Rakhit resistente "" síndrome saludable Fanconi.
Formas y patogénesis
Se distinguen dos tipos de síndrome de Fanconi:
- hereditario (congénito, idiopático), que se refiere a la forma primaria;
- adquirido, que se considera como una forma secundaria de la enfermedad.
Los especialistas en tipos hereditarios (genéticos) están asociados con un defecto X-cromosoma, que se hereda por tipo dominante y recesivo, por lo tanto, el pronóstico genético de su manifestación de futuras descendientes no es una tarea simple. Si la patología se relaciona con un tipo congénital (forma primaria), entonces se detecta en el niño en el período de pecho. Por lo tanto, la forma principal se llama "infantil".
El grado de mutación genético determina el grado de severidad del propio síndrome. Por lo tanto, el hereditario lleno de síndrome de Fanconi se descubre en presencia de 3 defectos bioquímicos básicos, que incluyen glucosuria, aminoaciduria, fosfantina, incompleta, con dos de ellos.
Típicamente, la patología determinada genéticamente está acompañada de otras enfermedades congénitas: cistinosis, síndromes de Wilson, Denta, baja, intolerancia a la fructosa, tirosinemia (la incapacidad del cuerpo dividido efectivamente el aminoácido de tirosina), galactosemia (disfunción de conversión de azúcar en glucosa), exceso Acumulación de glucógeno.
Desarrollo del síndrome de Fanconi.
El síndrome adquirido (secundario), en contraste con congénito, no está acompañado, pero es una consecuencia de las patologías existentes:
- tyrosiemia tipo I tipo;
- custodia (violación del metabolismo del aminoácido quístico con daño renal posterior);
- intolerancia a la fructosa;
- enfermedad de Wilson-Konovalov;
- galactosemia;
- glicogenesis (acumulación anormal de glucógeno en tejidos y órganos) tipo XI;
- patologías de riñón hereditarias;
- (violación del metabolismo de la proteína, lo que resulta en esclerosis, atrofia, disfunción orgánica);
- interestide de la tubula (jade con daño tisular, canales de riñón);
- hiperparatiroidismo ( enfermedad endocrinaen el que se perturbe el contenido normal en la sangre del calcio y el fósforo);
- educación malignada: mieloma, pulmón, páncreas, pulmones, cadenas ligeras,;
- quemaduras profundas.
Además, el síndrome puede provocar tales estados:
- trasplante del órgano a baja compatibilidad del tejido;
- escasez de vitaminas D;
- envenenamiento de uranio, bismuto, mercurio, plomo, cadmio;
- contacto con tolueno, ácido maleico, lysol;
- el uso de agentes farmacológicos nefrotóxicos, tales como: gentamicina, preparaciones de platino, fármacos basados \u200b\u200ben tetraciclina, didanosina, cdidopovir, medicamentos de quimioterapia contra el cáncer, iphosfamida, estreptozocina.
Síntomas y signos
En forma hereditaria (congénita)
Los síntomas primarios se manifiestan en los primeros meses de vida, rara vez después de un año y medio.
En primer lugar, el bebé recién nacido nota los siguientes estados:
- micción frecuente (poliuria);
- sed elevado (polidipsia);
- estreñimiento largo;
- ataques frecuentes de vómitos no dummados;
- astenia (fatiga total), debilidad muscular;
- temperaturas inexplicables de "saltos" hasta 37.5 - 38 c;
- pista hinchada.
Como regla general, en el período en que comienzan los combates de vómitos y los elevadores de temperatura del niño se muestran al pediatra. Un especialista experimentado debe determinar que la combinación de los padres preocupados de los signos no tiene relación con las infecciones ARS, ORVI o ENTEROVIRUS.
Un médico de niños competente es capaz de reconocer el síndrome de Fanconi a tiempo. PERO investigación de laboratorio - Confirmar sospechas Si se revelan tres (o dos) señales de base: glucosuria, hiperámicoidelum generalizado e hiperfosfatería característica de esta patología.
Después de síntomas de poca altura y suficientemente inciertos en el próximo año, uno y medio, los síntomas del síndrome de síndrome peculiar están claramente arreglados:
- Naíse temprano (ligereza) causado por la eliminación constante del organismo de aminoácidos esenciales, glucosa, calcio, fosfatos. Los primeros seis meses de crecimiento y peso normal se reemplazan por un déficit de peso corporal (hasta un 30%) y crecimiento (de 2 a 21%).
- Rahit, debido a la eliminación masiva de calcio y fosfatos, se hace notable después de 10 a 12 meses de vida, y tiene características características del síndrome de Fanconi: la cabeza del bebé suele ser un poco deformada, pero los huesos grandes de las piernas y las asas muestran curvaturas significativas: tensión En el tipo VIUS, cuando el niño que brilla, estamos torcidos con la "Rueda", o el Valgus (en forma de la letra "X"). Huesos de pecho, columna vertebral.
- Retraso en el desarrollo mental y físico.
- Comprensión, insugio, puesta en marcha.
- La polidipsia y la poliuria pueden progresar y retroceder, sin pasar finalmente.
- La hipotensión muscular moderada, que se expresa en la cámara lenta, la desventaja de los movimientos, lo que lleva al hecho de que los niños de 5 a 6 años no son capaces de caminar.
- Dolor de huesos intensidad moderadaMinding un niño para caminar. Mostrar más alto en el nivel de piernas, pelvis y columna vertebral. Gait, si un niño camina, se convierte en pato, inseguro.
- La alta probabilidad de fracturas de los huesos tubulares debido al déficit de minerales en el tejido óseo.
- Osteomalacia o suavizante de los huesos debido a la destrucción del tejido óseo como resultado de la falta de sales de calcio y fósforo.
- Reducción de la protección inmune a las infecciones, que se manifiesta en frecuentes. enfermedades virales, Otitis, Neumonía.
- Paraliamientos causados \u200b\u200bpor una escasez de potasio.
- Patologías oftalmológicas, tales como: pigmento retinit, catarata congénita.
- El desarrollo de patologías. sistema nervioso, ENT (oreja, nariz, garganta, laringe) y gastroy. del sistema cardiovascular, anomalías anatómicas de los órganos del sistema urinario debido a trastornos metabólicos masivos.
- En casos aislados - trastornos endocrinos
Al progreso de los trastornos tubulares (violación de tráfico sustancias orgánicas, minerales, electrolitos) por 10 a 12 años en niños han aumentado la probabilidad de lo crónico insuficiencia renalEso amenaza su vida.
Síntomas visibles del síndrome de Fanconi en niños. 
En pacientes adultos en el desarrollo de la forma secundaria.
Si los fankoni adquiridos por síndrome se desarrollan en personas de adultos con otras enfermedades o condiciones patológicasSus manifestaciones a menudo se combinan con las manifestaciones de la enfermedad del provocador.
Sin embargo, se han identificado señales básicos:
- Mayor volumen de orina por día (hasta 2 litros o más) y sed aguda, característica de los pacientes con la primera infancia.
- Debilidad general y muscular, dolor en los huesos.
- Alta probabilidad de la presión arterial obstinada () en el fondo de la disfunción del riñón.
- Osteomalacia (destrucción de los huesos).
- Acidosis (aumento de la acidez en la sangre) debido al retraso de los productos de oxidación en el cuerpo, lo que lleva a la hipopotasemia (deficiencia de potasio).
- Ophbromóticos: alta deposición de sales de calcio calcio con fiebre característica de este estado, frío, náuseas, dolor más fuerte en el abdomen, ingle, ovarios.
- Hipopotasemia (bajo flujo de potasio), causando complicaciones cardíacas severas, incluyendo vida amenazadora arritmia.
- Formación rápida (en ausencia de tratamiento) insuficiencia crónica riñón
Entre las mujeres
La versión más desfavorable del flujo de síndrome de Fanconi se implementa en su desarrollo en mujeres en el período de posmenopausia. En este momento, contra el fondo de la reducción de los productos hormonales, se produce una disminución natural en la densidad del tejido óseo (osteopenia).
Con una combinación de este estado con un aumento en la fragilidad ósea debido a la falta de minerales, la probabilidad de fracturas de compresión pesada de vértebras, cuellos de cadera y discapacidad posterior son altos.
Diagnóstico
Para exponer o refutar el diagnóstico, los huesos y las pruebas bioquímicas en profundidad de la sangre y la orina se llevan a cabo con la ayuda de la hentografía.
Laboratorio
Revelados cambios en la orina y la bioquímica de la sangre:
| Señales | Indicadores |
|---|---|
| contenido de calcio bajo y fósforo. | menos de 2.1 mmol / L y 0.9 mmol / L, respectivamente |
| acidosis ("con vistas a la sangre) | Ve \u003d 10 - 12 mmol / l |
| glucosuria (aumento del azúcar en la orina) | 2 - 3% y más |
| humenociduria (eliminación con orina importantes aminoácidos alanina, arginina, glicina, derrame) | hasta 2 - 2.5 g / día |
| remoción de calcio con orina | 1.5 - 3.5 mmol / día |
| aumento de la orina de pH (acidez) debido a la pérdida anormalmente alta de bicarbonato | hasta 6.0 |
| aumentar la densidad relativa de la orina. | 1,025 – 1,035 |
Durante el examen, también detecta:
- un aumento en la actividad de la fosfatasa alcalina;
- excreción excesiva del cuerpo de sales de sodio, potasio;
- proteinuria (la aparición de proteínas en la orina) en presencia de cadenas ligeras de inmunoglobulinas, lisozima, proteínas de bajo peso molecular, beta 2-microoglobulinas;
- mejora de la liquidación (tasa de filtración) de ácido úrico con su contenido de suero reducido
- la disminución de la actividad de las enzimas de intercambio de energía: sucibe deshidrogenasas, a-glicopelosfato deshidrogenasa, glutamato deshidrogenasa;
- aumentado en la sangre de la cantidad de ácido lácteo y piruogrado.
Instrumental
El diagnóstico del síndrome de Fanconi proporciona el uso obligatorio de la radiografía ósea para identificar la deformación del esqueleto, las extremidades, detectar signos de osteomalisis, osteoporosis y en niños, además, retrasos en el crecimiento óseo en comparación con la norma por edad calendario .
Se observan las siguientes anomalías en el tejido óseo:
- fibra gruesa, estructura celular con lacunas, mineralización débil, crecimientos anormales en forma de "espigas" en huesos femorales y bertov;
- síntomas de la epifisilación (parada parcial o completa del crecimiento óseo en un esqueleto largo, lo que lleva a la asimetría de las extremidades);
- fracturas de huesos tubulares (en la etapa tardía) contra los antecedentes del desarrollo de la osteoporosis, cuyo grado está determinado por la sustitución de rayos X;
- acumulación de radioisótopos en los campos de áreas intensivas de crecimiento óseo.
En los riñones:
En un estudio electrónico de la biopsia del tejido renal (biopsia), se detecta un cambio característico en la forma de los túbulos en forma de cuello de cisne, adelgazamiento, atrofia (disminución del volumen) del tejido epitelial en presencia de un Mayor cantidad de mitocondrias en ella, fibrosis (crecimiento anormal) del tejido conectivo.
Investigación relacionada:
Diferencial
La patología debe distinguirse de todas las enfermedades aisladas que puedan provocar la aparición del síndrome de Fanconi adquirido adquirido por estados e intoxicación insalubres.
Además, en lactantes, un pediatra está obligado a diferenciar la condición de la falta aguda de la vitamina D durante el síndrome de Fanconi desde su exceso de oferta al utilizar aditivos artificiales o trastornos de intercambio de calcio.
Diferencias en la hipervitaminosis D y Síndrome de Fanconi en niños hasta el año:
| Parámetros | Hipervitaminidad D. | Síndrome de Fanconi |
|---|---|---|
| Frecuencia | A menudo | Raramente |
| Síntomas (similares) | Piel seca, palidez, sed aguda, vómitos, larga estreñimiento, peso de peso y crecimiento, aumento del hígado | |
| Síntomas (diferenciándose) | Hipertensión (elevadores de presión arterial frecuentes) | Agotamiento, poliuria, hipotensión muscular, alta presión sanguínea Sin sangre |
| Sangre | Exceso de calcio en el período agudo. Contenido de fósforo reducido. Fosfatasa alcalina, azúcar, proteína - Normal | El calcio es más a menudo normal (puede ser reducido). Glucosa, proteína reducida. El fósforo se reduce bruscamente. La actividad de la fosfatasa alcalina se incrementa considerablemente, en 2 - 3 veces. Se levanta la acidez de la sangre |
| Orina | La prueba Sulkovic para el calcio es positiva. La presencia de proteínas, sangre (más de 2 a 3 glóbulos rojos en el campo de visión), leucocitos. Azúcar Aminoazot, como regla, es normal. | La prueba Sulcovich es negativa. Proteínas, Fosfatos, Sugar Elevado, AminoAciduria |
| Huesos | Las zonas de vigilancia están expandidas, selladas. | Osteoporosis de huesos tubulares, deficiencia de calcio en regiones ordinarias. |
Especialistas comprometidos en el síndrome de Fanconi y sus complicaciones: nefrólogo, ortopedia, hematólogo, endocrinólogo, urólogo, oftalmólogo, genética.
Tratamiento
La terapia racional y bien reflexiva es capaz de reducir el efecto en el cerebro, el sistema óseo y los órganos de la pérdida excesiva de aminoácidos, minerales, glucosa y proteína que se derivan de la orina.
Por lo tanto, el tratamiento con síndrome está dirigido a las siguientes tareas:
- La corrección máxima posible de la deficiencia de potasio, los bicarbonatos, los cambios en el equilibrio ácido-alcalino de la sangre para reducir la acidosis.
- Terapia de fosfato-diabetes (raquitis resistentes a D) con énfasis en la inadmisibilidad de la limitación de líquidos.
- Tratamiento de la enfermedad subyacente provocando el desarrollo del síndrome adquirido en adultos.
Medicinar
 Para mitigar el fósforo y las pérdidas de calcio. preparaciones especiales Con vitamina D, es L, 25 (OH) D3 y L (OH) D3.
Para mitigar el fósforo y las pérdidas de calcio. preparaciones especiales Con vitamina D, es L, 25 (OH) D3 y L (OH) D3.
Dosis primarias de vitamina D3 por día de 10 a 15 mil metros. Aumentar la dosis se realiza gradualmente, lo que aumenta cada 12 a 14 días (bajo el control de muestras de Sulkovich y el contenido de la sangre en fósforo). En ausencia de signos de intoxicación y una pequeña eliminación de calcio con la orina, se permite aumentar la dosis, aportar a 100-150 mil metros por día, y continuar con la terapia para indicadores normales En la sangre de fósforo y fosfatasa alcalina. Al estabilizar sus valores, la dosis no debe ser más.
La terapia con vitamina D se lleva a cabo por varios cursos para evitar crisis de progresión de las deformaciones de richitas en los huesos.
La opción óptima se considera que utiliza metabolitos activos D3 - OxideVIT (0.5 - 1.5 μg por día), Calciotryol (Rokallytrol).
Incluye preparaciones de calcio (gluconato de calcio por día a 1,5 - 2 gramos), fósforo (0,5 - 1 gramo por día), Fitin.
De los fosfatos inorgánicos, se usa una mezcla de Albright, llevándola a 1 cuchara grande 4 - 5 veces al día. Los fosfatos se utilizan en forma de una solución y tabletas en dosis de 10 mg por kilogramo de peso, 4 veces al día (obligatorio con preparaciones de vitamina D para evitar hiperparatirointerías).
Con una pronunciada escasez de potasio, panangina, asparks, se utilizan.
Con cualquier cita, el estado de la sangre de latón o ácido base se monitorea constantemente. Normalmente, la sangre tiene una reacción débilmente alcalina, y el pH en el rango de 7.35 - 7.45. Cuando la acidosis, cuando el pH se reduce por debajo de 7.35, la sangre adquiere una mayor acidez.
En estos casos, se muestra una infusión intravenosa de solución de hidrocarbonato del 4% o una solución de solución (50 a 60 ml por día) en la que ácido de limón - 2 gramos, citrato de sodio - 3 g, citrato de potasio - 3,3 g, 100 ml de agua. A 1 ml de dicha medicina de peeling contiene 1 mmol sodio y potasio. La alta acidez en la sangre se neutraliza también con refresco (bicarbonato de sodio).
Algunos especialistas en función de los resultados prácticos recomiendan Unitiol como un medio para aumentar la actividad de las enzimas tiológicas.
Se prescribe Customin: DITHIOTRENTAL a la velocidad de 25 mg por kilogramo del peso del paciente después de 3 horas; CISTEAMINE B. dosis diaria A la velocidad de 90 mg / kg.
Existen resultados positivos del uso de la penicilamina, lo que reduce la concentración de ácido peyrobico en la sangre, lo que reduce el grado de eliminación de los aminoácidos y contribuyendo al crecimiento de las reservas alcalinas.
Una buena influencia es trabajar por el anabólicos hormonales del canal de riñón, incluida la metiltestosterona.
El tratamiento en el hospital se muestra en trastornos metabólicos claramente pronunciados, incluidas la hiperglucemia hipo y la hiperglucemia, las deformaciones esqueléticas.
Prevención
Prevención moderna síndrome congénito Fanconi en presencia de tal patología en la familia consiste en consultoría genética preliminar. El riesgo de desarrollar la enfermedad para los llamados SIB, es decir, hermanas y hermanos, alrededor del 25%.
En la forma secundaria de su síndrome de síntomas, se reduce o desaparece completamente con el tratamiento activo de la enfermedad principal o la condición patológica adquirida.
Pronóstico
El pronóstico de la enfermedad depende del síndrome de forma (primaria, secundaria), severidad de las manifestaciones y el inicio del tratamiento.
Por ejemplo, los síntomas del síndrome adquirido desaparecen al eliminar la causa-provocador.
Para patología congénita Contra la ausencia de depósitos de cistina en los tejidos, el curso de la enfermedad no tiene una amenaza grave de vida, de lo contrario, especialmente, sin tratamiento adecuado, la muerte del paciente se proyecta a 10 a 20 años a partir de la creciente deficiencia de los riñones.
Sin embargo, incluso con cambios severos en los riñones: pielonefritis, nefritis interstal tubular, insuficiencia renal, con impacto temprano de medicamentos en proceso patológico Y la terapia a largo plazo establece un cierto equilibrio en la homeostasis, en el que el pronóstico de la calidad de vida normal durante décadas es bueno.
En la práctica médica hay enfermedades de la enfermedad, cuando los niños tienen 7-8 años, el síndrome de Fanconi hereditario prácticamente "se detuvo" con el inicio de la remisión a largo plazo, mejorando explícitamente la condición del niño e incluso la recuperación.
Síndrome de Debra - Debe - Fanfoni es una enfermedad innata grave, caracterizada por una variedad de niños del primer año de vida que la mayoría de ellos sufre. Como regla general, se encuentra junto con otras patologías hereditarias, pero puede manifestarse y como un síndrome independiente.
Breve excursión en la historia.
La enfermedad estaba abierta y estudiada en 1931 por el Dr. Fanconi de Suiza. Toperando a un niño con raquitis, bajo crecimiento y cambios en los análisis de orina, concluyó que esta combinación de signos debe considerarse como una patología separada. Dos años más tarde, De Tony hizo sus enmiendas, agregó a la descripción ya existente de hipofosfatems, y después de algún tiempo, Debe reveló de pacientes similares con aminoaciduria.
En la literatura doméstica, esta condición se llama los términos "Síndrome hereditario de Tony - Debre - Fanconi" y "glucoaminophosfatdiabet". En el extranjero se conoce más a menudo como síndrome de Fanconi Renal.
Causas del desarrollo del síndrome de Fanconi.
Actualmente, no fue posible averiguar completamente cuál era la base de esta enfermedad severa. El síndrome de Fanconi supuestamente es experto cree que el desarrollo de esta patología está asociado con una mutación puntual, que conduce al trabajo incorrecto de los riñones. Numerosos estudios confirmaron que en el cuerpo hay una violación del metabolismo celular. Es posible que el trifosfato de adenosina (ATP) esté involucrado en el caso, un compuesto que desempeña un papel importante en el intercambio de energía. Como resultado del trabajo incorrecto de las enzimas, la glucosa, los aminoácidos, los fosfatos y otras sustancias no menos útiles se pierden. En tales ambientes hostiles, los túbulos renales no reciben energía necesarios para su trabajo. Material útil se describen con la orina, violada procesos de intercambio, Los cambios rahitoides en el tejido óseo se están desarrollando.

El síndrome de Fanconi en niños es mucho más común en adultos. Según las estadísticas, la frecuencia de la patología es de 1: 350,000 recién nacidos. Los niños y las niñas en iguales proporciones están enfermas.
Signos de síndrome fankoni
La enfermedad puede desarrollarse a cualquier edad, pero con mayor frecuencia se celebra en niños del primer año de vida. Glucosuria, hiperámico generalizado e hiperfosfatio: esta tríada de signos caracteriza el síndrome de Fanconi. Los síntomas se desarrollan bastante temprano. En primer lugar, los padres notan que su hijo comienza a orinar con más frecuencia, y atormenta constantemente la sed. Los niños, por supuesto, no pueden decir sobre esto con las palabras, pero en su comportamiento caprichoso y su colgante constante en el pecho o una botella, se vuelve obvio que algo está mal con el niño.
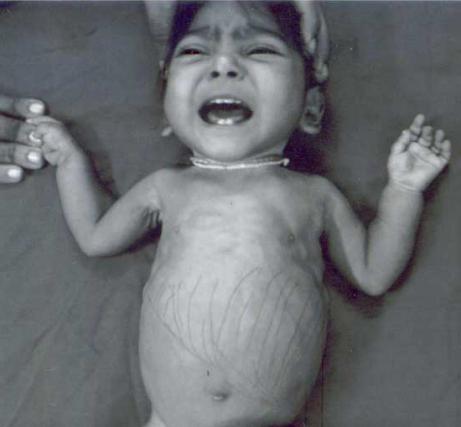
EN más padres Trae mucha preocupación de que el vómitos indebidos frecuentes, un largo estreñimiento y no se explican como una regla, en esta etapa, el niño finalmente cae al médico. Un pediatra experimentado puede sospechar que una combinación de síntomas no es como un resfriado convencional. Si el médico es competente, podrá reconocer el síndrome de Fanconi a tiempo.
Mientras tanto, los síntomas no desaparecen en ninguna parte. Añaden un retraso destacado en físico y desarrollo mentalAparecen curvaturas severas de huesos grandes. Usualmente los cambios afectan solamente miembros inferiores, lo que lleva a la tensión en el tipo variante o Hughus. En el primer caso, las piernas del niño se torcerán con la rueda, en la segunda, en forma de la letra "X". Y la otra opción, por supuesto, desfavorable para la vida futura del niño.
El síndrome de Fanconi en niños a menudo incluye osteoporosis (destrucción prematura del tejido óseo), así como un aumento significativo en el crecimiento. No se excluyen las fracturas largas y paralizales. Incluso si es así, los padres no han preocupado por el estado del bebé, en esta etapa definitivamente no rechazarán la asistencia calificada.

El síndrome de Fanconi en adultos es bastante raro. La cosa es que esta enfermedad grave naturalmente conduce al desarrollo de la insuficiencia renal. Con esta situación, es imposible dar cualquier pronóstico inequívoco y garantizar una gran cantidad de casos descritos en la literatura cuando, a la edad de 7 a 8 años, el síndrome de Fanconi aprobó su posición, hubo una mejora notable en la condición del niño e incluso la recuperación. . Desafortunadamente, tales opciones en la práctica moderna son lo suficientemente raras como para hacer conclusiones serias.
Diagnóstico del síndrome de Fanconi.
Además de recopilar anamnesis y inspección cuidadosa, el médico definitivamente asignará algunas encuestas para confirmar esta enfermedad. El síndrome de Fanconi inevitablemente conduce a una violación del trabajo de riñón, lo que significa que será el análisis de orina habitual. Por supuesto, esto no es suficiente para identificar todas las características del curso de la enfermedad. Es necesario ver no solo el contenido en la orina de proteínas y leucocitos, sino que también intente detectar la lisozima, las inmunoglobulinas y otras sustancias. En el análisis, también será revelado. alto contenido El azúcar (glucosa), los fosfatos (Phosphautour), será visible para pérdidas significativas de sustancias importantes para el cuerpo. Dicha encuesta puede llevarse a cabo ambos pacientes ambulatorios como en el hospital.

En los análisis de sangre, algunos cambios también son inevitables. Para estudio bioquímico Existe una disminución en casi todos los microelementos significativos (principalmente calcio y fósforo). Desarrollado por un pronunciado interfiero trabajo normal Organismo entero.
La radiografía del esqueleto mostrará la osteoporosis (destrucción del hueso) y la deformación de las extremidades. En la mayoría de los casos, se encuentra el retraso de la tasa de crecimiento de los huesos y su incumplimiento de la edad biológica. Si es necesario, el médico puede asignar riñones de ultrasonido y otros órganos internos, así como una encuesta de especialistas relacionados.
Diagnóstico diferencial
Hay casos en que algunas otras enfermedades están enmascaradas en el síndrome de Fanconi. Antes de que el doctor se levante tarea difícil Para averiguar qué sucede con un pequeño paciente de hecho. A veces la glucoaminophicepiabetabet está confundida con pielonefritis crónica y otras enfermedades renales. Cambios en las pruebas de orina, así como características Las lesiones óseas ayudarán al pediatra a establecer el diagnóstico correcto.
Tratamiento del síndrome de Fanconi.
Vale la pena considerar el hecho de que esta patología es crónica. Es bastante difícil deshacerse completamente de los síntomas desagradables, solo es posible durante algún tiempo reducir las manifestaciones de la enfermedad. Lo que sugiere medicina moderna ¿Como ayudar a los niños enfermos?
En primer lugar hay una dieta. Los pacientes recomiendan limitar sales, así como a todos los productos nítidos y ahumados. Leche y varios jugos dulces de frutas se agregan a la dieta. No te olvides de (ciruelas, secas y pasas). En el caso de que el déficit de los oligoelementos alcanzó la etapa crítica, los médicos prescriben una recepción de complejos de vitaminas especiales.
En el contexto de la dieta, se introducen grandes dosis de vitamina D. La condición del paciente se monitorea constantemente, tiene que donar sangre y orina de vez en cuando. Esto es necesario para revelar el hipervitaminidad inicial en el tiempo y reducir la dosis de vitamina D. Tratamiento a largo plazo, grandes cursos, con interrupciones. En la mayoría de los casos, dicha terapia ayuda a restaurar el metabolismo perturbado y prevenir complicaciones severas.

Si la enfermedad ha ido lejos, el paciente cae en manos de los cirujanos. Los ortopedistas experimentados podrán enderezar las deformaciones óseas y aumentar significativamente el nivel de vida del niño. Dichas operaciones se realizan solo en el caso de un bastidor y remisión a largo plazo: al menos un año y medio.
Pronóstico
Desafortunadamente, el pronóstico para tales pacientes es desfavorable. En la mayoría de los casos, la enfermedad avanza lentamente, tarde o temprano, lo que lleva a la insuficiencia renal. Las deformaciones de los huesos del esqueleto conducen inevitablemente a la discapacidad y el deterioro de la calidad general de la vida.
¿Es posible evitar esta patología? Sin lugar a dudas, tal pregunta le preocupa a todos los que han encontrado el síndrome de Fanconi. Los padres están tratando de entender lo que no lo hicieron y dónde no se descartan. Es igualmente importante saber si la repetición de la situación con otros niños está amenazada. Desafortunadamente, actualmente no se desarrollan medidas de prevención. Los parames que planean para lograr que otro niño se consulte en Genética para obtener más información completa sobre el problema emocionante.
Síndrome de Viserler - Fanconi (subsiptación alérgica)
Esta enfermedad se describe solo en niños de 4 a 12 años. La razón de esta patología seria aún no se conoce. Se puede suponer que este síndrome es típico. una enfermedad autoinmune, forma especial de artritis reumatoide. Siempre comienza de manera aguda, con un elevador de temperatura, que se puede mantener en la cifra de 39 grados. En todos los casos, la erupción polimórfica aparece en las extremidades, a veces en la cara, el pecho o el estómago. Por lo general, la recuperación, sin complicaciones graves. Sin embargo, la parte de los pacientes pequeños a tiempo desarrolla lesiones graves de las articulaciones que conducen a la discapacidad.
Síndrome de Fanconi (más correctamente del síndrome de Tony-Debrey-Fanfoni): la disfunción generalizada de los túbulos proximales, que son los siguientes trastornos (incluidos los descritos): 1) acidosis canálica proximal con bicarbonateuria; 2) glucosa renal; 3) Phosphautour; hipofosfatemia; raquitismo hipofosfatémico; 4) Hipostenuria (poliuria); 5) aminoaciduria; 6) Proteinurium de tipo tubular (cadenas de inmunoglobulina ligera, proteínas de bajo peso molecular - microglobulina P2). Además, se observa la pérdida de sodio, potasio, calcio, acumulación de acumulación de ácido úrico con una disminución en el contenido del suero.
Causas del síndrome fankoni
El síndrome de Fanconi puede ser una enfermedad primaria (hereditaria o adquirida), es más a menudo secundaria, desarrollando bajo un número enfermedades comunes. La causa del síndrome de Fanconi puede ser trastornos metabólicos hereditarios (cistinosis, galactosemia, enfermedad de Vilson-Konovalov); envenenamiento sustancias toxicas (por ejemplo, salicilato, tetraciclina con un caducado) y metales pesados \u200b\u200b(plomo, cadmio, bismuto, mercurio); neoplasias malignas (mieloma, enfermedad de las cadenas ligeras, cáncer de ovario, hígado, pulmón, páncreas); Linfogranulomatosis. El síndrome de Fanconi también puede desarrollarse en algunas enfermedades de los riñones, con hiperparatiroidismo, hemoglobinuria de noche paroxística, quemaduras pesadas.
Signos clínicos del síndrome de Fanconi.
Signos clínicos Invita a la derrota de los huesos (deformación esquelética, dolor óseo, fracturas, osteomalacia difusa), los niños desarrollan raquitis, retraso en el crecimiento. Puede haber poliuria, sed, rara vez la debilidad muscular (hasta la parálisis), asociada con la hipopotasemia, las convulsiones hipocalcémicas. Los niños redujeron la resiliencia a las infecciones. Los signos clínicos pueden estar ausentes, el diagnóstico en tales casos se basa en datos de laboratorio que detectan la violación integrada de las funciones tubulares.
A pesar de que en formas hereditarias, aparecen los primeros signos en infanciaLa enfermedad a veces se reconoce a una edad mayor. No hay signos clínicos ni de laboratorio que distinguen el síndrome de Fancoon primario de la secundaria, por lo tanto, en cada caso, se debe realizar una búsqueda etiológica completa.
El tratamiento se lleva a cabo mediante grandes dosis de bicarbonatos, se prescriben mezclas de citrato, vitamina D, preparaciones de potasio, una dieta de papa-col-col.

