La insuficiencia selectiva de IGA es el estado de inmunodeficiencia primaria más común (PIDS). La capacidad de los pacientes con deficiencia de IgA selectiva varía de 1: 400 a 1: 1000 en una población similar a una población europea y significativamente más baja, de 1: 4000 a 1: 20,000, en el mongoloide. En los Estados Unidos, la prevalencia de la enfermedad varía de 1 a 223-1000 en el grupo estudiada hasta 1 a 400-3000 en donantes de sangre saludables. En Rusia, tales estudios no se realizaron.
Para este estado, la disminución selectiva en la concentración sérica de la IGA se caracteriza por debajo de 0.05 g / L (en niños durante cuatro años) nivel normal Otras inmunoglobulinas séricas, reacción normal Los anticuerpos séricos y una respuesta inmune indirecta con células convencionales. En la mayoría de los estudios, la frecuencia de ocurrencia entre los representantes masculinos y femeninos fue aproximadamente la misma.
Las personas con la incapacidad de producir IgA pueden llevar su enfermedad asiméticamente debido a mecanismos de compensación o sufrir infecciones respiratorias frecuentes, digestivas o buen sistema, patología gastroenterológica (por ejemplo, enfermedad celíaca), una tendencia a trastornos atópicos, como la poliniasis, asma bronquial, Dermatitis atópica, igé-indirect alergia a la comida, así como enfermedades neurológicas y autoinmunes (la mayoría de las veces es la artritis reumatoide, el lupus rojo sistémico, el púrpura trombocitopénico idiopático, el síndrome de SheGreen). Con insuficiencia selectiva igA. enfermedades alérgicas, como la dermatitis atópica y el asma bronquial, se reunieron en el 40% de los casos (Consilium Medicum, 2006). También característica de la mayoría de los pacientes reacciones anafilácticas al rebosar componentes sanguíneos y administración de inmunoglobulinas intravenosas, que se asocia con la presencia de IGA en estos productos.
Los síntomas clínicos de la deficiencia selectiva de la IGA pueden manifestarse en la primera infancia, con la edad de la misma frecuencia y la gravedad de las infecciones portátiles pueden disminuir debido al aumento compensatorio de los anticuerpos de los subgradios IgG1 y G3, IgM. Otra explicación de la ausencia. síntomas clínicos Puede haber un nivel normal de IGA secretor, a pesar de la reducción en el nivel de inmunoglobulina sérica. O, por el contrario, algunos pacientes con una deficiencia de IGA selectiva inicialmente diagnosticados pueden desarrollar la clínica con una falla inmune variable común.
La terapia de la deficiencia selectiva de la IGA se encuentra actualmente en la identificación de enfermedades concomitantes mer preventivorayas Para reducir el riesgo de infección, así como el tratamiento rápido y eficaz de las infecciones.
No hay tratamiento específico. El pronóstico en pacientes con deficiencia de IgA es generalmente bueno, si no se expresa manifestaciones clínicas. La insuficiencia de la IGA en los niños se puede alimentar con el tiempo.
Al ser determinados genéticamente, los estados de inmunodeficiencia surgen debido a los defectos del aparato genético. Los pacientes con insuficiencia inmune variable común y aquellos con deficiencia de IgA selectiva a menudo se detectan en una familia y tienen un HLA-haplotipo común; Muchos tienen alelos raros y eliminaciones de genes dentro de la clase 3 de la clase 3 en el cromosoma 6. Recientemente se ha demostrado que algunos casos familiares de la insuficiencia inmunitaria variable total y la deficiencia selectiva de la IGA son causadas por la mutación del gen TNFRSF13B, que codifica la proteína conocida como Taci (activador transmembrana y modulador de calcio y interactor de ciclofilina-ligando). Es probable que en los casos en que no se encontraron mutaciones taci, la causa de las enfermedades podría servir como mutaciones espontáneas o hereditarias de otros genes, aún no fijados.
Actualmente, posibles manifestaciones clínicas de deficiencia selectiva de IgA, opciones de flujo, posibles enfermedades concomitantes se describen bastante detalladamente. Decisión en el diagnóstico de la enfermedad es la disminución selectiva de la concentración sérica de la IGA en niños de 4 años por debajo de 0.05 g / L en el nivel normal de otras inmunoglobulinas séricas en inmunogramas repetidos. El tratamiento consiste en identificar enfermedades concomitantes, realizando medidas preventivas para reducir el riesgo de infección, y también se necesita rápido y tratamiento efectivo enfermedades infecciosas.
La información sobre la frecuencia de ocurrencia de este estado primario de inmunodeficiencia en la población rusa está ausente, lo que no permite comparar la prevalencia de la enfermedad en nuestro país con otros países, donde estos estudios ya se han realizado.
El principal problema es la ausencia de recomendaciones uniformes sobre las tácticas de pacientes con deficiencia selectiva de IgA.
Para evaluar la frecuencia de ocurrencia de la deficiencia selectiva de la IGA entre los niños del grupo. observación de dispensarios "Frecuentemente enfermos" y caracterizan el espectro de sus manifestaciones clínicas en Federación Rusa Sobre la base de FGBU "FNCC Dgoo nombrado después de Dmitry Rogachev" Ministerio de Salud de la Federación de Rusia y GBUZ DGKB No. 9. G. N. Speransky Dzz se realizó este trabajo.
Materiales y métodos de investigación.
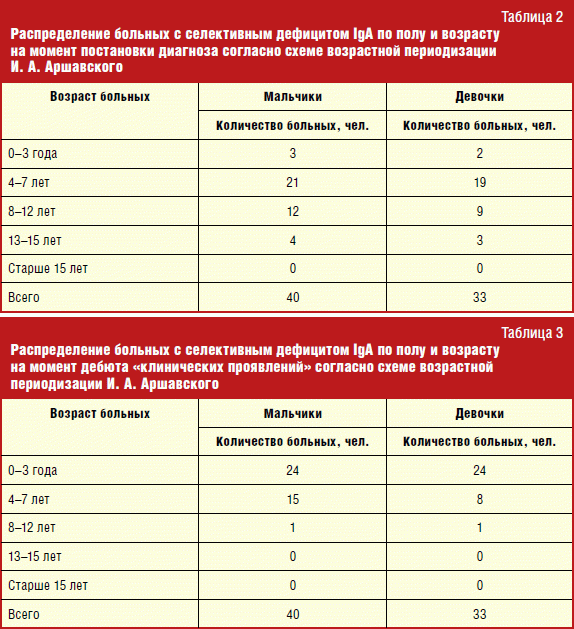
Los niños con deficiencia selectiva de IGA, observados en GBUZ DGKB No. 9 se han convertido en un objeto de estudio. G. N. SPERANSKY DZZ. Además, analizó retrospectivamente los registros médicos para el período de 2003 a 2010. 9154 Pacientes del grupo de observación de dispensarios "Niños más enfermos" (Tabla 1-3).
Durante el examen, se utilizaron los siguientes métodos:
- clínico y anamnésico;
- general I. análisis bioquímicos sangre;
- estudio inmunológico de la composición de la sangre mediante métodos de aceite de aceite y citometría de flujo;
- muestras de escarificación;
- determinación de un método específico de IgE de inmunotransferencia;
- estudio de la función de la respiración externa;
- investigación rinocitológica.
El diagnóstico de deficiencia selectiva de la IGA se elevó sobre la base de una disminución selectiva en la concentración sérica de IGA por debajo de 0.05 g / L indicadores normales Otras inmunoglobulinas séricas en los inmunogramas repetidos y la exclusión de los demás. posibles Causas Su insuficiencia para niños mayores de 4 años.
Al recoger anamnesis atención especial Frecuencia y espectro pagados de manifestaciones clínicas, patología concomitante, así como la historia familiar estudiada en detalle. El examen clínico de los niños se realizó de acuerdo con los métodos generalmente aceptados. El contenido de las inmunoglobulinas de las clases A, G, M, E en el suero se determinó mediante el método de medidor de aceite en el medidor de aceite BN 100 (Dade Bering, Alemania) utilizando Dade Behring Set. El fenotipado de los linfocitos se llevó a cabo mediante el método de citometría de flujo en el dispositivo FACSSCAN (Becton Dickenson, EE. UU.) Usando los anticuerpos monoclonales marcados con fluorescencia simultánencia (Becton Dickenson, EE. UU.). Pacientes con manifestaciones de atopy, así como a todos los pacientes con nivel de aumento IgE, que se identificó como resultado de un resultado estimado del estado inmune por el método de medidor de aceite, se llevó a cabo la adicción alergológica mediante el método de las pruebas de escarificación en niños mayores de 4 años o determinando la IGA específica en el suero en pacientes menores de 4 años. años de edad. Los niños con un diagnóstico de "asma bronquial" o la presencia del síndrome de bronucho-prestical en la historia realizó un estudio de la función de la respiración externa en el aparato SPIROVIT SP-1 (Schiller AG, Suiza). Todas las direcciones y consultas necesarias de especialistas relacionados, teniendo en cuenta las quejas disponibles.
Los resultados y su discusión
El análisis retrospectivo de los registros médicos de los pacientes con diagnósticos de guía "Recurient ARVI", "CHBD", "CBD", así como "EBD", permitió establecer que la frecuencia de la deficiencia selectiva de la IGA en este grupo de niños es dos. , e incluso tres veces más alto que en la población.
El número absoluto, así como el porcentaje de niños con esta inmunodeficiencia primaria, se puede ver en la tabla. cuatro.
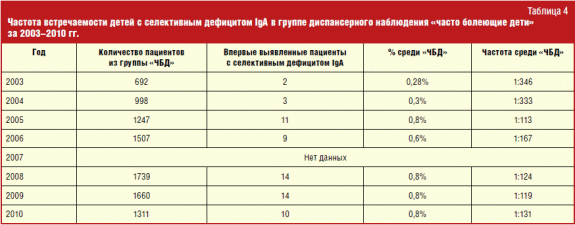
Desafortunadamente, los datos de 2007 no están disponibles. En 2003 y 2004 692 y 998 niños fueron consultados. Entre ellos se encontraban un total de 5 pacientes con deficiencia selectiva de IgA, que es un poco más frecuente que en promedio por la población: 1: 346 y 1: 333, respectivamente, contra 1: 400-600. A partir de 2005, la frecuencia por primera vez identificó a los pacientes con este PID, aumentó dramáticamente: 1: 113 en 2005, 1: 167 en 2006, 1: 124 en 2008, 1: 119 en 2009 y, finalmente, 1: 131 en 2010. Durante el estudio, la frecuencia de ocurrencia cambió de 1: 346 en 2003 a 1: 131 en 2010, cuando resultó ser máximo en comparación con los años anteriores. Aumentar la frecuencia de aparición de pacientes con deficiencia de IgA selectiva para el tercer año después del inicio del trabajo debe asociarse con el mayor estado de alerta de los médicos en relación con esta patología, además de mejorar diagnóstico de laboratorio. Es necesario continuar expandiendo el conocimiento de los médicos sobre esta enfermedad, ya que el flujo de niños que los padres llevan a un inmunólogo con quejas sobre enfermedades frecuentes, aumenta de año a otro.
En el curso de este trabajo, 235 niños y 32 adultos también fueron encuestados prospectivamente.
El grupo principal fue de 73 niños con una deficiencia selectiva diagnosticada de IGA.
El segundo grupo de pacientes incluyó a 153 niños con púrpura trombocitopénica idiopática (ITP). La evaluación del estado inmune a los pacientes con ITP se llevó a cabo para identificar la deficiencia de IGA selectiva entre ellos, ya que esta correlación se describió en la literatura mundial y se obtuvieron los mismos datos durante este estudio. Ni un solo hijo con la falta de IgA entre ellos no se identificó. A pesar de que durante el examen del estado inmunológico de los pacientes con ITP, no pudimos identificar la deficiencia selectiva de la IGA entre ellos, se identificaron otros pequeños defectos humorales: la deficiencia de la subclase IgG, la hipogammaglobulinemia infantil, la disminución parcial de la IGA.
El tercer grupo incluyó a 32 adultos de 20 a 54 años, así como 8 niños de 4 a 10 años, que son los parientes más cercanos de los pacientes con deficiencia selectiva de IgA, que se evalúa el estado inmunológico para buscar y describir los casos familiares. .
Durante la encuesta y el análisis de los datos obtenidos, se obtuvieron los resultados descritos a continuación.
La proporción de hombres y mujeres entre pacientes con deficiencia selectiva de IGA fue aproximadamente la misma. Se examinaron 40 niños y 33 niñas. Esto corresponde a la literatura mundial.
El pico de la detección de la deficiencia selectiva de la IGA representa los 4 a 7 años de edad. Las repetidas enfermedades infecciosas, como regla general, surgieron en temprana edad O con el comienzo de una visita a la institución preescolar de los niños. Como regla general, antes de llegar al inmunólogo, los niños acumularon una cierta historia infecciosa, ya que hay ciertos signos que les permiten sospechar los PID. Y, además, incluso si el estudio se realizó a una edad más temprana y la ausencia de IGA se reveló a 4 años, no nos permitió poner un diagnóstico inequívoco de PID, no pudimos excluir completamente la inmadurez del sistema de síntesis. de inmunoglobulinas. Por lo tanto, hasta 4 años, el diagnóstico se exhibió sobre temas y se recomendó la observación en la dinámica. De ahí el intervalo de 4-7 años, respectivamente.
Las principales quejas al manejar a los niños con deficiencia de IgA selectiva fueron frecuentes respiratorios. infecciones virales con flujo sin complicaciones. Las enfermedades respiratorias recurrentes de débito, como regla general, representaron edades de hasta 3 años. También corresponde a la literatura mundial. Dado que el control dinámico sobre la mayoría de los pacientes de nuestro estudio se llevó a cabo durante mucho tiempo, durante varios años, a veces antes de la transición del paciente en la red adulta, se puede argumentar que con la edad, la frecuencia y la gravedad de las infecciones portátiles disminuyeron. Se suponía que se debía al aumento compensatorio de los anticuerpos de la Subclase IgG1 e IgG3, IgM, pero esta pregunta requiere un estudio adicional. El segundo en frecuencia es una queja al aplicar fue una orvisitud frecuente que fluye con complicaciones. La frecuencia de complicada, atípicamente realizada por Orvi con la edad en nuestros pacientes, como se muestra observación dinámica, también disminuyó.
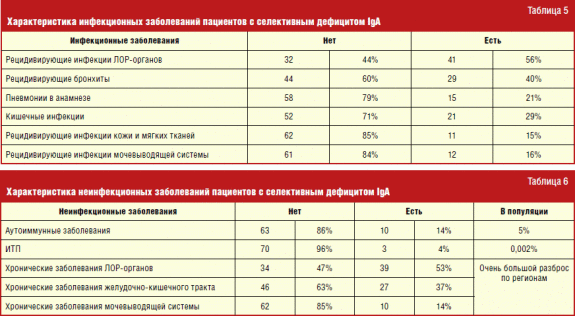
Desde el espectro de enfermedades infecciosas en pacientes con deficiencia selectiva de IGA, el lugar líder fue ocupado por enfermedades infecciosas de los órganos ENT e infecciones más bajas. tracto respiratorio. Esto se debe al hecho de que la disminución en la IGA secretora, que forma parte de la inmunidad local, conduce a la infección ligera y la reproducción de microorganismos en las membranas mucosas, más vulnerables a los contactos con enfermedades infecciosasTransmitiendo gotitas en el aire.
En el espectro de enfermedades no transmisibles, se reveló una correlación obvia con enfermedades autoinmunes, que son las manifestaciones más importantes de la deficiencia selectiva de la IGA, en particular, con un púrpura trombocitopénico idiopático (1.5-2 por 100 mil).
De enfermedades autoinmunes En pacientes con deficiencia selectiva de IgA, la artritis reumatoide juvenil (4 veces), la púrpura trombocitopénica idiopática crónica (3 veces), la hepatitis autoinmune (3 veces) fue la más común. Además, según la literatura mundial, en pacientes con deficiencia selectiva de la IGA, se observa una mayor frecuencia de los estados autoinmunes entre los parientes más cercanos. Pero, según nuestra investigación, su número no excedió los valores de generalización.
La frecuencia de las enfermedades atópicas entre los pacientes con deficiencia de IgA selectiva resultó ser significativamente más alta que en la población (Tabla 4). Solo la frecuencia de la rinitis alérgica es comparable al idioma general. Dichas observaciones se reflejan en una serie de estudios previamente realizados. No se puede decir que las enfermedades alérgicas en la mayoría de los pacientes con deficiencia de IgA sean más difíciles que en los humanos sin este defecto inmunológico. Sin embargo, la mayor prevalencia de Atopia da una razón para plantear la cuestión de realizar un examen inmunológico para identificar las formas de deficiencia selectiva de la IGA, que, aunque clínicamente no se mostraron. Aunque puede que no tenga un papel decisivo en términos de enfoque de la terapia para el estado atópico actual, pero lo ayudará a realizar un diagnóstico de manera oportuna y reducirá los posibles riesgos para las personas que tengan una deficiencia selectiva de IgA.
Al analizar los inmunogramas repetidos durante la observación dinámica en los niños con deficiencia selectiva de IgA, en relación con los cambios persistentes en los indicadores de laboratorio, se asignaron dos dos grandes grupos pacientes. En el grupo A, no hubo IgA sin ningún otro cambio. El grupo en ausencia de IgA se combinó con un aumento persistente en la IgG. Se realizó un análisis comparativo de estos grupos de pacientes.

La edad del debut de las manifestaciones clínicas en estos grupos no fue confiable.
Se reveló que en pacientes con deficiencia selectiva de IgA, un aumento en el nivel de IgG se correlaciona con enfermedades infecciosas repetidas de la piel y los tejidos blandos. Esta pregunta requiere un estudio adicional.
Al comparar estos grupos de pacientes, no se detectaron diferencias significativas en el espectro de alergopatología.
En el curso del trabajo, se realizó una evaluación del estado inmunológico en 20 familias de pacientes con deficiencia selectiva de IGA. Se revelaron 4 casos familiares. Además, se recolectó una historia familiar detallada. Entre los familiares adultos con una historia infecciosa cargada que logró realizar una encuesta, había esos u otros deterioro de la inmunidad humoral. En consecuencia, al identificar pequeños defectos humorales (en particular, la deficiencia selectiva de la IGA), el examen de los familiares más cercanos, especialmente en presencia de una anamnesis infecciosa cargada, es obligatorio.
Debido al hecho de que la deficiencia selectiva de la IGA entre los hijos del grupo de observación de dispensarios "a menudo enfermos" es mucho más común que en una población de niños comunes, practicando los pediatras necesitan ser cautelosos con respecto a esta enfermedad. No siempre es fácil sospecharlo, ya que las manifestaciones clínicas son muy variables: de las formas asintomáticas a infecciones bacterianas recurrentes con la necesidad de frecuentes terapia antibacteriana. Se recomienda ampliar el conocimiento de los pediatras y los especialistas en pacientes ambulatorios y estacionarios estrechos sobre los pequeños defectos de la inmunidad humoral.
Dado que entre los pacientes con deficiencia de IgA selectiva son significativamente más altas que la frecuencia de la patología alérgica (asma bronquial, dermatitis atópica, alergias a los alimentos), por encima de la frecuencia de las enfermedades autoinmunes y las enfermedades hematológicas, así como la frecuencia. enfermedades crónicas (Órganos enteros, sistema urogenital, tracto gastrointestinal) que en la población, su identificación es obligatoria con el fin de proporcionar un pleno derecho y su temporal atención médica Pacientes.
Se recomienda que se recomienda consultar a un inmunólogo / examen inmunológico de los niños con anamnesis infecciosa hidratada, pacientes con enfermedades hematológicas y autoinmunes, para realizar encuestas de detección del nivel de pacientes generales de IGA con la presencia de enfermedades alérgicas.
En el curso del estudio, se estableció que la mayoría de los niños con déficit selectivo de IGA marcaban la correlación. patología autoinmune y un aumento persistente en IgG en inmunogramas repetidos. Con otras enfermedades, tal correlación no se estableció. Los cambios similares en los indicadores son un factor de riesgo en la patología autoinmune del niño y requieren atención especial.
A pesar del hecho de que las correlaciones entre la presencia de la historia de la historia familiar de la deficiencia selectiva de la IGA y la gravedad de las manifestaciones clínicas en pacientes no se establecieron, para estos pacientes, examen de los parientes más cercanos, especialmente en presencia de una anamnesis infecciosa cargada. , es obligatorio.
Literatura
- Hammarstrom L., LONNQVIST B., Ringden O., Smith C. I., Wiebe T. Transferencia de deficiencia de IGA a un paciente injertado de médula ósea con anemia aplásica // Lancet. 1985; 1 (8432): 778-781.
- Latiff A. H., Kerr M. A. A.La importancia clínica de la inmunoglobulina una deficiencia // análisis de la bioquímica clínica. 2007; 44 (PT 2): 131-139.
- Al-Attas R. A., Rahi A. H.Deficiencia de anticuerpos primarios en los árabes: Primer informe de la Arabia Saudita / / Journal de Inmunología Clínica. 1998; 18 (5): 368-371.
- Carneiro-Sampaio M. M. M., Carbonelare S. B., Rozentraub R. B., De Araujo M. N., Riberiro M. A., Porto M. H.Frecuencia de la deficiencia selectiva de la IGA entre los donantes brasileños de sangre y las mujeres embarazadas sanas // Alergología inmunopatología (MADR). 1989; 17 (4): 213-216.
- Ezeoke A. C. Deficiencia selectiva de IGA (SIGAD) en el este de Nigeria // Revista africana de medicina y ciencias médicas. 1988; 17 (1): 17-21.
- Feng l. Estudio epidemiológico de la deficiencia selectiva de IGA entre 6 nacionalidades en China // Zhonghua Yi Xue ZA ZHI. 1992; 72 (2): 88-90, 128.
- Pereira L. F., Sapina A. M., Arroyo J., Vinuelas J., Bardaji R. M., Prieto L. Prevalencia de deficiencia selectiva de IGA en España: más de lo que nosotros / / sangre. 1997; 90 (2): 893.
- Wiebe V., Helal A., Lefranc M. P., Lefranc G. Análisis molecular de la eliminación de Multigene Multigene del T17 Inmunoglobulin CH (DEL A1-GP-G2-G4-E) // Genética humana. 1994; 93 (5): 5.
L. A. A. Fedorova *,
E. S. Pushkova *
I. A. KORSUNSKY **, 1, Candidato de Ciencias Médicas
A. P. PRODEUS *,doctor en Ciencias Médicas, Profesor.
Inmunodeficiencia - Indicadores cuantitativos reducidos y / o actividad funcional de los componentes principales sistema inmune, lo que lleva a una violación de la protección del cuerpo de los microorganismos patógenos y se manifiesta por una mayor incidencia infecciosa.
Como se sabe, la función principal del sistema de inmunidad es el reconocimiento y la eliminación de sustancias extrañas de la naturaleza antigénica que penetra en el cuerpo de ambiente (microorganismos) o arisco endógeno (células tumorales). Esta característica se implementa utilizando factores. inmunidad congénita (Phagocitosis, péptidos antimicrobianos, proteínas del sistema de complemento, sistemas de células NK, etc.) y adquiridas, o adaptables inmunes, llevadas a cabo utilizando respuestas inmunes celulares y humorales. La regulación de la actividad de los componentes de la protección inmunitaria del organismo y su interacción se producen con la ayuda de citocinas y contactos intercelulares.
En cada uno de los componentes listados del sistema inmunológico, así como en los mecanismos de su regulación, pueden ocurrir trastornos, que conducen al desarrollo de la inmunodeficiencia, cuya manifestación clínica principal es una mayor sensibilidad a los agentes causales de enfermedades infecciosas. Dos tipos de inmunodeficiencia distinguen: primaria y secundaria.
Inmunodeficiencia primaria(PID) - enfermedades hereditariasCausado por defectos de los genes que controlan la respuesta inmune. PID: enfermedades, diversas en la naturaleza y severidad de los defectos inmunes, manifestaciones clínicas y trastornos moleculares. Para la imagen clínica, el PID se caracteriza por procesos infecciosos graves, repetidos y crónicos, en mayor grado del sistema broncopulmonar.
y órganos enteros, piel y membranas mucosas; Se pueden desarrollar linfadenitas purulentes, abscesos, osteomielitis, meningitis y sepsis. En algunas formas hay manifestaciones de alergias, enfermedades autoinmunes y el desarrollo de algunos tumores malignos. Deberías prestar atención al atraso. indicadores de edad desarrollo fisico. Actualmente, se describen aproximadamente 80 PID, los genes responsables del desarrollo de la mayoría de estas enfermedades. Las pruebas de laboratorio adecuadas permiten diferenciar la patología a nivel de linfocito y patología a nivel de destrucción no celular y mecanismos de eliminación de antígenos.
Prevalencia de piddepende de la forma de la enfermedad y en el rango promedio de 1: 10,000 a 1: 100,000 recién nacidos. Déficit selectivo IgA, por ejemplo, ocurre mucho más a menudo de 1: 500 a 1: 1500 personas de la población general. Predominio varias formas Pid varía en diferentes paises. La mayoría de las veces, hay defectos de los anticuerpos: 50-60% de los casos, PID combinados - 10-30%, defectos de fagocitosis - 10-20%, defectos de complemento - 1-6%. La mayoría de los PID se manifiestan en la primera infancia, aunque es posible ser mejor ser el comienzo de algunas formas de PID, en particular, la falla inmunológica variable total (OVIN).
Para los mecanismos de desarrollo, se asignan 4 grupos PID principales:
1er grupo - predominantemente humoral, o en células
Pid;
2do Grupo - PID combinado (con todos los inmunodeficiadores de células T, hay una violación de la función de las células B);
El 3er grupo - PID, debido a defectos de fagocitosis;
El 4º Grupo - PID, debido a los defectos en el sistema de complemento.
Principios de diagnóstico de inmunodeficiencia primaria.
El diagnóstico precoz y el inicio oportuno del tratamiento determinan el pronóstico de la enfermedad. La formulación del diagnóstico a nivel de los pediatras del distrito presenta ciertas dificultades, lo que a menudo se debe a la ausencia de la posibilidad de consultar oportunamente a un paciente con un médico-inmunólogo y realizar un examen inmunológico de laboratorio especial (Tabla 11-1). Aunque el conocimiento de las características de la imagen clínica PID y el cambio.
en las pruebas de laboratorio clínico general, le permite sospechar PID y enviar pacientes a los expertos en la técnica. La sociedad europea de inmunodeficiencia ha desarrollado protocolos para el diagnóstico temprano de PID, y también creado base electrónica Datos del Registro Europeo PID. El algoritmo de diagnóstico PID se muestra en la FIG. 11-1.
Tabla 11-1.Etapas de examen inmunológico con sospecha de inmunodeficiencia.
Higo. 11-1.Algoritmo para diagnósticos de inmunodeficiencia primaria.
Características generales de la imagen clínica de la inmunodeficiencia primaria.
Dirigido B. cuadro clinico Pid es el llamado síndrome infeccioso - Mayor susceptibilidad a los agentes causales de enfermedades infecciosas en general, inusualmente recurrentes graves (recurrentes) corriente clínica, La presencia en la etiología de la enfermedad de patógenos atípicos (a menudo oportunistas). El tipo de patógeno está determinado por la naturaleza del defecto inmune. Con defectos de anticuerpos, es posible identificar firme drogas antibacterianas Floer - Staphylococci, Streptococi, varita hemofílica. Con falla inmune de células T, además de las bacterias, los virus revelan (por ejemplo, una familia de herpesvirus), hongos (Candida spp., Aspergilluset al.), y con defectos fagocíticos - Stafilococos, bacterias gramnegativas, hongos, etc.
Investigación de laboratorio
Si los datos clínicos le permiten sospechar PID, se deben realizar los siguientes estudios:
La determinación de la fórmula de sangre desplegada (los indicadores cuantitativos y porcentuales de los linfocitos son especialmente importantes);
Definición de niveles IgG, IgA y IgM en suero sanguíneo;
Cálculo de las subpoblaciones de los linfocitos T-y B;
◊ Análisis del estado funcional de los fagocitos (el análisis más simple e informativo es una prueba para la restauración del azul tetrazolium);
◊ Análisis sobre el contenido de los componentes principales del complemento (inicio con C3 y C4);
◊ Análisis para la infección por VIH (si es posible, factores de riesgo);
◊ Estudios genéticos moleculares cuando testimonio.
Principios de tratamiento de la inmunodeficiencia primaria.
El objetivo principal de la terapia PID es el tratamiento de las complicaciones de la enfermedad y su prevención. Este enfoque es causado por el hecho de que los defectos del sistema inmunológico con PID se colocan a nivel genético. Actualmente realiza investigaciones intensivas sobre gen.
terapia de ruido de la inmunodeficiencia, que puede llevar a la aparición de métodos más radicales de su tratamiento.
Dependiendo de la forma del PID, el tratamiento es realizar la terapia de sustitución, el tratamiento y la prevención de las manifestaciones autoinmunes infecciosas de la enfermedad, el tratamiento de neoplasias malignas y la aplicación métodos especiales, incluido el trasplante de células madre hematopoyéticas (dependiendo del tipo de PID).
Defectos de inmunoglobulina
Hipogammaglobulinemia transitoria en niños
La hipogammaglobulinemia transitoria en niños está asociada con una característica fisiológica de la formación gradual del sistema de inmunoglobulina. La maduración de la formación de anticuerpos IgM y IgA es la mayoría de los "retrasos". En niños sanos, el contenido de la IgG materna disminuye gradualmente y después de medio año aumenta el desarrollo de sus propios anticuerpos IgG. Sin embargo, en algunos niños, se retrasa el aumento en el nivel de inmunoglobulinas. Tales niños pueden sufrir enfermedades infecciosas bacterianas recurrentes. En estos casos, no debe recurrir a las infusiones de las drogas de las inmunoglobulinas de los donantes (introducción de inmunoglobulina intravenosa).
Deficiencia selectiva de inmunoglobulina a
Inmunoglobulina A (SD IGA - Deficiencia selectiva de IgA)desarrollado como resultado de un defecto genético. tNFRSF13B.
o p). La deficiencia de IGA en presencia de inmunoglobulinas de otras clases es la inmunodeficiencia más frecuente detectada en una población común con una frecuencia de 1: 500-1500 personas (en pacientes que sufren alergias, más a menudo). Insuficiencia selectiva de IGA, es decir, Contribuyendo en una escasez de una de las subclases (30% de los casos) y completa (70% de los casos). La deficiencia de la subclase IgA2 conduce a una imagen clínica más pronunciada que el déficit de la subclase IgA1. Las combinaciones de deficiencia de IgA con otras violaciones son posibles: con defectos de biosíntesis de IgG y anomalías de linfocitos T. La gran mayoría de las personas con selectividad.
la deficiencia de la IGA es prácticamente saludable. Para niños menores de 2 años, la deficiencia de IgA es un estado fisiológico.
Revelar la reducción de la concentración sérica de IgA a<5 мг/дл у детей старше 4 лет; IgG и IgM в норме, количество и соотношение субпопуляций лимфоцитов и их функциональная активность могут быть в норме.
Cuadro clinico.Con una deficiencia de IgA, se pueden desarrollar 3 grupos de síndromes patológicos: infecciosos, autoinmunes y alérgicos. Los pacientes con deficiencia de IgA están predispuestos a enfermedades infecciosas recurrentes del tracto respiratorio superior y los órganos digestivos. Los más frecuentes y gravemente fluyen son una variedad de enfermedades autoinmunes (artritis reumatoide, spondilitis anquilosante, síndrome de granos, vasculitis con una lesión de vasos cerebrales, tiroiditis autoinmune, LES, glomerulonefritis, anemia hemolítica, diabetes tipo I, vitiligo, etc.). La frecuencia de Celiachood excede tales en niños con IgA normal 10 veces. Las manifestaciones alérgicas más detectadas: la intolerancia a la proteína de la leche de vaca, la dermatitis atópica (ATD), el asma bronquial.
Tratamiento.Los casos de flujo asintomático no requieren ningún tratamiento especial; En presencia de manifestaciones clínicas de enfermedades infecciosas, autoinmunes y alérgicas, el tratamiento se realiza de acuerdo con las normas.
La terapia de reemplazo de las inmunoglobulinas de los donantes no se muestra en selectiva, ni con una deficiencia completa de la IGA, ya que la probabilidad de la formación de anticuerpos anti-etióticos a la IGA y el desarrollo de complicaciones de transfusión causadas por ellos es alta.
Agamaglobulinemia con deficiencia de células B
Aghamaglobulinemia agarrada X (enfermedad de Bruton)es el 90% de todos los casos de Aghamaglobulinemia. Los niños están enfermos, hijos (אּ, ρ) de portadores de un gen defectuoso bTK (XQ21.3-Q22),proteinthyrosinkinasa proteinthyrosinkinasa B-Lymphocyte BTK (Bruton "s tirosina quinasa- Bruton tirosina quinasa). Como resultado del defecto, una violación de las trayectorias de señalización intracelular, la recombinación de las cadenas de inmunoglobulina pesadas, difunto
recuperación de células BR B en los linfocitos B. En el 10% de los pacientes con agammaglobulinemia con deficiencia en células, se hereda la autosomongénesis. 6 Los defectos genéticos se describen actualmente, incluidas las moléculas del receptor de células pre-B, las células B del adaptador citoplásmico (BLNK) y el gen Rico en leucina que contiene 8 (LRRC8).
Datos de laboratorio.No hay linfocitos B periféricos. En la médula ósea hay células pre-B con una cadena μ en el citoplasma. El número de linfocitos T y las pruebas funcionales en los linfocitos T pueden ser normales. IgM y IgA en la sangre no pueden ser revelados; IgG puede estar presente, pero en pequeñas cantidades (0.4-1.0 g / l). No hay anticuerpos contra los antígenos de los grupos sanguíneos y los antígenos de la vacuna (tétanos, toxinas difterias, etc.). La neutropenia puede desarrollarse. Estudio histológico de la tela linfoide: no hay centros germinativos (germinales) y células plasmáticas en los folículos linfoides.
Cuadro clinico.Si el historial familiar es desconocido, el diagnóstico se vuelve obvio en promedio durante 3,5 años. La enfermedad se caracteriza por un tejido linfoide de hipoplasia, infecciones purulentas que fluyen pesadamente, enfermedades infecciosas de la parte superior (sinusitarios, otitis) y menor (bronquitis, neumonía) del tracto respiratorio; Gastroenteritis, piodermia, artritis séptica (bacteriana o clamazdiosa), septicemia, meningitis, encefalitis, osteomielitis. Como agentes causales de enfermedades respiratorias, la mayoría de las veces se realiza. Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus,bacterias intestinales de la diarrea o Giardia Giardia Lamblia.Además, los pacientes con Aghamaglobulinemia son susceptibles a enfermedades infecciosas causadas por micoplasmas y ureasmos que causan el desarrollo de neumonias crónicas, artritis purulenta, cistita y absceso subcutáneo. Desde virus, virus neurotrópicos típicos ECHO-19 y cokes, causando tanto la encefalitis pesada y crónica y la encefalomielitis. Las manifestaciones de las infecciones de enterovirus pueden ser un síndrome de dermatomía, una ataxia, dolores de cabeza, violaciones de comportamiento. En pacientes con niños en inmunización, un poliocresino vivo, como regla general, se detecta una liberación a largo plazo del virus de la polio a través de las membranas mucosas, y con la virulencia restaurada y creciente (es decir, en el colector de niños.
somos un peligro real de infección de niños sanos con polio, como resultado del contacto con un niño inmunodeficiente vacunado). Los trastornos autoinmunes durante la agammoglobulinemia pueden representarse por artritis reumatoide, síndrome de esclerodermo, escleroderm, colitis ulcerosa no específica, diabetes tipo I (debido a la predominio de la respuesta inmune TH1).
Inspección física.Preste atención al retraso en el desarrollo físico, en la forma de los dedos (dedos en forma de palos de tambor), cambios en la forma del tórax, característicos de las enfermedades del tracto respiratorio inferior, la hipoplasia de los ganglios linfáticos y las almendras.
Tratamiento.
Terapia de reemplazo: las fármacos de inmunoglobulina intravenosa se introducen cada 3-4 semanas para la vida. Las dosis de inmunoglobulinas se seleccionan para crear en el suero del paciente su concentración, superponiendo al límite inferior de la norma de edad.
Discutir la posibilidad de terapia génica - gen BTK.se cloneó, sin embargo, su hiperexpresión está asociada con una transformación maligna del tejido hematopoyético.
En el caso de la neutropenia persistente, se utilizan factores de crecimiento. Con la aparición de signos de patología autoinmune, es posible designar medicamentos de anticuerpos monoclonales (infliximab, etc.).
Fracaso inmune variable general
La falla inmune variable común (OVIN) es un grupo de síndromes caracterizados por un defecto para la síntesis de anticuerpos y la inmunidad celular. El criterio de diagnóstico confiable de AVIN es una reducción significativa en el contenido de las inmunoglobulinas de dos o tres isotipos principales en personas de ambos sexos en combinación con uno de los siguientes signos:
Debut de la enfermedad mayor de 2 años;
Falta de isohemaglutininas y / o baja respuesta de vacunación;
Una exclusión de otras razones de Aghamglobulinemia.
En algunos pacientes, la causa del desarrollo de AVIN es la mutación de los genes que codifican moléculas involucradas en los procesos de maduración y supervivencia de las células B: BAFF-R (Receptor de factor de activación de células B)BLIMP-1. (Proteína de maduración inducida por linfocitos B)y iCOS. (Costimulador inducible).Existe una violación de la capacidad B-Lymphocyte para diferenciarse en células plasmáticas, se están desarrollando defectos de anticuerpos, se está desarrollando disfunción en T linfocitos, se observa una mayor tendencia a las enfermedades infecciosas. El síndrome puede manifestarse en la primera infancia, en la adolescencia o en los jóvenes.
Datos de laboratorio.Los niveles de IgG e IgA se reducen significativamente (aproximadamente el 50% de los pacientes) y IgM (hasta la discusión). El número de linfocitos B en la sangre corresponde a la norma o reducido. El número de linfocitos T en la mayoría de los pacientes es normal. En pacientes severos, el desarrollo de la linfopración es posible (menos de 1500x10 3 células en 1 litro de sangre). El número de células NK se reduce. La producción de anticuerpos específicos en respuesta a la inmunización se reduce o ausente. La proliferación de linfocitos y la formación de IL-2 bajo la acción de mitógeno y antígenos se violan significativamente.
Cuadro clinico.Reclutar enfermedades infecciosas bacterianas recurrentes con localización predominantemente en el tracto respiratorio y los senos incompletos. En el momento del diagnóstico de la infección respiratoria, el tracto respiratorio puede progresar a la bronquiectasia y las lesiones derramadas de tejido claro. Es posible un daño infeccioso a los órganos digestivos, manifestados por la diarrea, el vapor y la malabsorción (y, en consecuencia, la pérdida del peso corporal). A menudo identifica infecciones causadas por Giardia Lamblia, Pneumocystis Cariniio virus familiares Herpetoviridae.Los pacientes de AVIV son propensos al desarrollo de la artritis purulenta causada por micoplasmas y ureapasmos. Las manifestaciones de las infecciones por enteravirus pueden ser la encefalomielitis, los síndromes polémicos y similares a la dermatomía, lesiones de la piel y las membranas mucosas. Autoinmunelas enfermedades ocurren duras y pueden determinar el pronóstico de OVIN. A veces, las primeras manifestaciones clínicas de AVIVE son la artritis, la colitis ulcerosa no específica y la enfermedad de Crohn, la colangitis esclerosante, la malabsorción, el LES, la nefritis, la miositis, el daño autoinmune a los pulmones en forma de neumonita intersticial linfoide, neutropenia,
púrpura trombocitopénica, anemia hemolítica, anemia perniciosa, alopecia total, vasculitis retina, fotosensibilización. En pacientes con ovina, la frecuencia aumenta significativamente (en el 15% de los casos), el gránulo similar a los sarcoidos y la linfoproliferación no maligna. Tratamiento.
Quimioterapia antobacteriana.
Terapia de reemplazo: las fármacos de inmunoglobulina intravenosa se introducen cada 3-4 semanas para la vida.
En las complicaciones autoinmunes, la terapia inmunosupresora (glucocorticoides, la azatioprina, la ciclosporina A) y es posible prescribir preparaciones de anticuerpos monoclonales (infliximab, etc.).
Síndromes de Hyper-IgM
Los síndromes de Hyper-IgM son enfermedades bastante raras caracterizadas por una disminución pronunciada o ausencia completa de IgG, IgA y una concentración de IgM en suero normal o elevada. Esto es causado por la incapacidad de los linfocitos B para cambiar las clases de inmunoglobulina y la hipermutagénesis de dominios variables. Hasta la fecha, se han identificado 6 defectos genéticos que llevan al desarrollo de Hyper-IGMSPROM.
. TIPO 1 (HIGM 1).Deficiencia de LIGAND DE LIGAND DE LIGNADA X (70% de los síndromes de Hyper-IgM), que conduce a la incapacidad de las células T para interactuar de manera efectiva en los linfocitos.
. Tipo 2 (HIGM 2).Autosomal-recesivo, asociado con un defecto de ayuda, la activación inducida por la citidina deamindaasa (gen AICIDA, 12P13)- Enzima involucrada en las clases de conmutación de inmunoglobulinas e hipermutagénesis.
. Tipo 3 (Higm 3).Autosomal recesivo, asociado con la mutación del gen de la molécula CD40. Al mismo tiempo, las células B en sí mismas no pueden interactuar de manera efectiva con los linfocitos T. Las manifestaciones fenotípicas son similares a las de tipo 1.
. Tipo 4 (HIGM 4).Autosómica recesiva; En algunos casos, surgen mutaciones. de Novo.Sonaba con un ung - uracil-dna-glicosyase - enzima, que también participa
en las clases de conmutación de inmunoglobulinas, pero después de la acción de ayuda. En este caso, la hipermutagénesis no afectará y el síndrome fluye con menor severidad.
. Tipo 5 (Higm 5).Defecto solo en las clases de conmutación, la hipermutagénesis no afectará. La mutación causal aún no se ha detectado, pero, obviamente, el defecto en la enzima actuando después de
AYUDA.
. TIPO 6 (HIGM-ED).X-alojado, asociado con una displasia ectodérmica de descarga, causada por una deficiencia de NEMO (modulador NF-KB), lo que lleva a una violación de transmisión de señal con CD40.
Síndrome de Hyper-IgM con Transferencia Xrevelar más a menudo. Desarrollado cuando un defecto de genes de codificación CD40L (CD154, el gen se encuentra en Xq26-q27.2)- Ligand para CD40. La insuficiencia de la expresión de los linfocitos T CD40L conduce a la imposibilidad de cambiar las clases de inmunoglobulina en los linfocitos B con IgM a otros isotipos, así como a una violación de la formación de células B de memoria, repertorio de células T y TH1. Respuesta celular dirigida contra microorganismos intracelulares. Los niños están enfermos
Datos de laboratorio.IgG, IgA, IgE no se puede determinar ni detectar en cantidades muy pequeñas. El nivel IGM es normal (en el 50% de los casos) o elevado, a menudo significativamente. La cantidad de células T y B es normal; Reducción de la respuesta proliferativa de las células T inducidas por los antígenos. IgM es policlonal, a veces monoclonal. Se detecta antena terapia del isotipo IgM (anti-aleatorio, antitrombocito, anti-aleatorio, anticuerpos a los antígenos de tejido muscular liso). En tela linfoide, no hay centros germinativos, pero hay células plasmáticas.
Cuadro clinico.Las primeras manifestaciones surgen en la infancia infantil y temprana. Característico repetido infecciónvarias localizaciones (principalmente tracto respiratorio), incluyendo oportunistas (causadas Pneumocystis carinii).También característica de los virus (citomegalovirus y adenovirus), Criptococcus neoformans,mycoplasmas y Mycobacteriums. La infección criptosporidial puede causar diarrea aguda y crónica (desarrollándose en el 50% de los pacientes) y la colangitis esclerosante. Anemia, neutropenia, ulceración de la mucosa oral, gingivitis, ulcerativa.
abejas del esófago, varios departamentos intestinales, colitis ulcerosa no específica. Revelar la predisposición a K. trastornos autoinmunes(Artritis seronegativa, glomerulonefritis, etc.) y neoplasias malignas (principalmente tejido linfoide, hígado y tracto biliar). Linfadenopatía, hepato y esplenomegalia pueden desarrollarse. Tratamiento
Terapia de reemplazo regular con inmunoglobulina intravenosa.
Quimioterapia antobacteriana. Para la prevención y el tratamiento de los neumonios neumónicos, se utilizan co-trimoxazol [sulfametoxazol + trimethoprim] y pentamidina.
Para evitar la lesión del hígado y el tracto biliar, solo se debe usar agua hervida o filtrada, examine regularmente (examen de ultrasonido, biopsia hepática de acuerdo con las indicaciones).
En el tratamiento de la neutropenia y las ulceraciones de la cavidad oral, se utilizan glucocorticoides y preparaciones de un factor de colonización de granulocitos.
En el desarrollo de complicaciones autoinmunes, se prescriben terapia inmunosupresora (glucocorticoides, azatioprina, ciclosporina A), así como medicamentos basados \u200b\u200ben anticuerpos monoclonales.
El método óptimo de tratamiento es el trasplante de médula ósea de los donantes compatibles con HLA (el nivel de supervivencia es del 68%, es mejor conducirse menor de 8 años).
Inmunodeficiencia combinada con predominante defecto T-Lymphocyte
Fallo inmune combinado pesado
Tkin (scid - Deficiencia inmunitaria combinada severa)- un grupo de síndromes caracterizados por una disminución en el nivel de los linfocitos T o su ausencia completa y su inmunidad adaptativa con discapacidad. . Disticular Disgénesis, caracterizada por una violación de la maduración de antecesores linfoides y mieloides en las primeras etapas: neutropenia y T - B - NK -.
. X-agarrado tkin, que se desarrolla como resultado de una mutación genética IL-2RG.[(CD132, TOTAL w.-Compra el receptor para IL-2, IL-4, IL-7, IL-9, IL-15 e IL-21), XQ13.1-Q21.1,אּ ], lo que conduce al bloqueo de los receptores y la incapacidad de las células diana para responder a la acción de las interleukinas correspondientes (más del 50% de todos los casos tkin); T - B + NK -.
. Falta de tirosina quinasa Janus3 [Gene Jak3 (19p13.1),ρ ]; cuando el gen defecta, la transmisión de la señal de activación del general. w.-Cake IL-2, IL-4, IL-7, IL-9, IL-15, IL-21 al núcleo de la célula, que conduce a una perturbación de la diferenciación de las células T- y NK; T - B + NK -.
. Insuficiencia de proteintithosinfosfatasa (CD45, Gene PTPRC, 1T31-Q32);cuando el defecto del gen se incrementa por la actividad inhibitoria de la CSK quinasa en la proteintetínidikinakinasa SRC con una violación de la fosforilación de los dominios de ITAM TCR y BCR; T - b + nk +.
. Deficiencia completa de las enzimas RAG1 y RAG2 que activan la recombinación V (D) de los segmentos J de inmunoglobulinas y TCR [Genes Rag1y RAG2 (11p13),ρ ]; T - b - nk +.
. El síndrome es Intenna (escasez incompleta de enzimas RAG1 y
Rag2) [genes Rag1y / o RAG2 (11p13-P12),r].Gracias
la baja actividad residual de estas enzimas todavía está desarrollando una serie de clones de linfocitos T específicos para los antígenos de los tejidos epiteliales de la piel y el tracto digestivo, donde multiplican y producen grandes cantidades de IL-4 e IL-5, causando hiperoeosinophyla. y la formación de linfocitos B residuales de IgE (en ausencia de inmunoglobulinas de otras clases). Caracterizar la eritrodermia y la pachidermia con la alopecia en el cuero cabelludo y las cejas, diarrea agotadora, síndrome infeccioso de la vida amenazadora; Hepatoslenomegalia y hiperplasia ganglios linfáticos.
. Tankin con mayor sensibilidad a la radiación ionizante. Defecto de la proteína nuclear Artemisa [gen Dclre1c, (10p),r],incluido en el complejo de las enzimas necesarias para la reparación de ADN (participa en el compuesto de roturas de doble cadena), con una mutación del gen, una violación V (d) J-recombinación; T - b - nk +.
. Deficiencia de IL-2 [Gene IL-2, 4T26-Q27].
Mutación del gen del receptor del receptor de IL-2 (CD25) (10p15-P14);T - b + nk +.
Mutaciones del gen del receptor del receptor de IL-7 (CD127) (5p13);T - b + nk +.
Deficiencia de alquitrán Transporter para la presentación del antígeno),péptidos antigénicos necesarios en el reticulum endoplásmico del gen del receptor de IL-7 (CD127) (5p13);T - b + nk +.
Las mutaciones de los genes de la cadena CD3 (CD3γ, CDδ y CDε), lo que llevó a una disminución en el número de linfocitos T maduros, la interrupción de su diferenciación; T - b + nk +.
Insuficiencia de proteinthirosinkinasa zap-70 [gen ZAP-70 (2T12),r]. En la mutación del gen, la fosforilación de las cadenas de ITAM ζ de TCR y los receptores de células NK que contienen ITAM están sufriendo, la deficiencia selectiva de las células T CD8 + está en desarrollo (el contenido de los linfocitos T CD4 + es normal, pero funcional Los trastornos se expresan en forma de la falta de educación por estas células 2 y la proliferación).
Deficiencia de formación de adenosina [gen ada (20Q12-Q13.11 , p)], lo que lleva a la acumulación en células de metabolitos (desoxiadenocrifosfato y s-adenosilgomocisteína), inhibiendo la proliferación de linfocitos T-y B (opciones para el debut tardío de la enfermedad); T - b - nk -.
Insuficiencia de la p[gen pNP (14T11.2),p], lo que lleva a la acumulación de desoxiiguanosintrifosfato en las células que inhiben la proliferación de los linfocitos T (síndromes acompañantes, Urrikamia y Urikuria); T - B + NK -.
Datos de laboratorio.Recluta variable, a veces linfopod profundo; Los linfocitos no pueden proliferar en respuesta a un antígeno específico; A menudo, a menudo se expresa una disminución en el nivel de inmunoglobulinas en el suero de sangre. No hay sombra de timo en la radiografía del tórax.
Cuadro clinico.Por lo general, el diagnóstico clínico se aclara en los primeros 6 meses de vida, cuando desaparecen los anticuerpos maternos IgG. En la imagen clínica en primer plano síndrome infeccioso severohipoplasia de tejido lirfero y retraso del desarrollo. El síndrome infeccioso se caracteriza por la candidiasis oral, la diarrea crónica, la neumonía, la fiebre,
septete de etiología bacteriana, infecciones virales. Infecciones de infecciones pertenecen a diferentes grupos taxonómicos: bacterias, virus, hongos, microorganismos condicionalmente patógenos. (Pneumocystis carinii).La neumonía es a menudo causada P. carinii,diarrea - Rotavirus, Campylobacter, Giardia Lamblia.A menudo manifiesta la hepatitis viral. Caracterizado por el desarrollo de BSZhit regional o generalizado después de la vacunación.
Tratamientoproporciona el nombramiento de la terapia de apoyo, incluida la nutrición parenteral, la introducción de la inmunoglobulina intravenosa, el propósito de los antibióticos, los antifúngicos y las drogas antivirales. Uno de los principales métodos de tratamiento para lograr la recuperación: el trasplante de médula ósea, sin los cuales los niños con tankina suelen morir en el 1er año de la vida. Los casos individuales se describen cuando el niño ha vivido en condiciones particularmente desinfectadas por hasta 2-3 años. Es importante reconocer que lo más pronto sea lo posible en los recién nacidos, en cuanto a ellos fatales, por ejemplo, la inmunización de las vacunas vivas. Inmediatamente después de establecer el diagnóstico de tales niños, es necesario poner en las condiciones no novelobiológicas (caja estéril). En el caso de la fijación de enfermedades infecciosas, la terapia intravenosa de reemplazo de la inmunoglobulina intravenosa, la terapia de reemplazo de inmunoglobulina intravenosa. Para la prevención de la neumonía neumática, se prescribe co-trimoxazol. En caso de desarrollo de BVZHIT, es necesario realizar una intensa terapia contra la tuberculosis a largo plazo. Para la transfusión de componentes sanguíneos, solo se deben usar medicamentos irradiados y filtrados. Existe el riesgo de una reacción posterior a la transfusión "trasplante contra el huésped" en relación con la transferencia transplacenta de los linfocitos maternos.
Síndrome "Linfocitos desnudos"
La llamada patología, cuando las moléculas MHC-I o MNS-II no se expresan en el cuerpo. En ausencia de expresión de las moléculas MHC-I, el contenido de los linfocitos T CD8 + se reduce y no hay actividad de las células NK; En ausencia de MNS-II, se reduce el nivel de los linfocitos T CD4 +. Se caracterizan algunos defectos genéticos. Sin embargo, estos defectos se localizan no en los genes MHC, sino en varios factores diferentes responsables de regularlos.
expresión. Cuadro clinicosíndrome de linfocitos "desnudos" y tratamientosimilar a aquellos con el resto de tiendas.
Síndrome de djori
En el síndrome de Djori, o el síndrome de defecto de defectos y el cuarto bolsillos faríngales [eliminaciones en 22Q11incluyendo gen TBX1 (22T11.2),revelan hipoplasia o aplasia de timo, hipoplásico de la glándula paracaída, deficiencia cardíaca, deficiencia de corazón, deficiencia de linfocitos T, cantidades variables de b-linfocitos.
Datos de laboratorio.Una disminución significativa en la cantidad de células T CD3 +, CD4 + y CD8 + y una disminución aguda en su actividad proliferativa inducida por mitógeno y antígenos. El número de células B- y NK es normal. La concentración de inmunoglobulinas séricas en la mayoría de los casos dentro del rango normal, son posibles varias opciones para la disbammaglobulinemia.
Cuadro clinico.El componente de inmunodeficiencia está representado por hipoplasia o armario de timo y recurrente, fluyendo severamente. enfermedades infecciosas.Higoparatiroidismo (hipocalcemia y, como resultado, - Totania, notable por 1-2 días después del nacimiento); Los vicios del sistema circulatorio (la inversión correcta del arco de la aorta, la estenosis del ventrículo derecho, los defectos en las particiones interventriculares e interpresentaciones, Tetrad Fallo, atresia o la helicoplascencia de la arteria de la luz); Wpadin nyaba; Anomalías del esqueleto facial (distancia aumentada entre los órganos emparejados, las mandíbulas de tamaño reducido, especialmente los sumideros de oreja de baja planificación, surcos de bandeja corta). Expresas anomalías de la estructura de la laringe, faringe, tráquea, oído interno, esófago; Violación del desarrollo de los riñones, SNC y otras malformaciones (polidactilación, falta de uñas, ANUS ARESIA, Fistulos anal). El retraso del habla y el desarrollo psicomotor es característico. Notas la predisposición a K. trastornos autoinmunes(citopenia, tiroiditis autoinmune) y neoplasias malignas.
Tratamiento.. Terapia antibacteriana y antiviral. . Terapia de reemplazo con inmunoglobulina intravenosa. . Tratamiento quirúrgico para el propósito de la corrección de malformaciones. . En complicaciones autoinmunes - terapia inmunosupresora. . Si hay endocrinopatías, corrección de trastornos apropiados. . Trasplante de médula ósea no efecto
tIVNE. . El trasplante de tejido epitelial TIMUS está justificado. Corrección de la función de los vidrios paracólogos.
Síndrome linfoproliferativo enganchado de X
X- el síndrome linfoproliferativo capturado se caracteriza por una violación de una respuesta inmune al virus de Epstein-Barr [debido a defectos genéticos Sh2d1a.(SAP) en Xq25אּ], lo que lleva a la proliferación incontrolada de los linfocitos B transformados por el virus Epstein-Barr, y la infección con el virus con nuevas células diana.
Cuadro clinico.Los 4 fenotipos más comunes se describen: la mononucleosis infecciosa severa, las condiciones linfoproliferativas malignas (linfoma, la leucemia, principalmente en la célula), la anemia o la pancing (incluyendo debido al síndrome de hemofagocítica inducido por el virus), la disbammaginemia. Infectando el virus de Epstein-Barr es un mecanismo de activación (lanzador) para la formación de las enfermedades más pesadas, en movimiento y fatales: la mononucleosis infecciosa fulminante (en el 58% de los casos conduce a la muerte), el síndrome de hemofagocítica (sin tratamiento en el 100% De los casos conduce a un resultado fatal). En el 10% de los casos, el fenotipo se manifiesta antes de la infección con el virus Epstein-Barr (la descarga de la disbammaglobulinemia y el linfoma se desarrollan). La mayoría a menudo detecta varios tipos de hipigammaglobulinemia. La inmunodeficiencia conduce al desarrollo de bacterianos, fúngicos y virales. enfermedades infecciosas.Puede asumir la enfermedad en niños con una historia familiar característica y se le atendió la prueba PCR positiva en el virus de Epstein-Barr. Para diagnosticar, se recomienda utilizar una combinación de análisis genético. Sh2d1a.y estimaciones del nivel de expresión de SAP.
Tratamiento
Con el objetivo de la prevención, se recomienda utilizar medicamentos antivirales - aciclovir, valacyclovir (su propósito temprano suprime la replicación del virus epstein-Barr en el rotoglotka y la inmunoglobulina intravenosa (con un título alto de anticuerpos al virus de Epstein-Barr).
En la hipogammaglobulinemia, la inmunoglobulina intravenosa se usa mensualmente en combinación con la terapia antibacteriana.
Se prescribe dosis altas de aciclovir y metilprednisolona para el tratamiento de la mononucleosis infecciosa fulminante, la terapia de alto grado con una inmunoglobulina intravenosa con un título alto de anticuerpos contra el virus Epstein-Barr y IFNA.
Con el desarrollo del síndrome de hemofagocítica combina dosis altas de dexametasona con una semezida ♠ (etopósido).
En el tratamiento de enfermedades malignas, se utilizan protocolos de terapia estándar.
Método de tratamiento radical: trasplante de médula ósea de los donantes compatibles con HLA.
Síndrome linfoproliferativo autoinmune
Síndrome linfoproliferativo autoinmune: un grupo de enfermedades caracterizadas por la linfoproliferación benigna, la hiperimmunoglobulinemia, los trastornos autoinmunes, un mayor contenido de CD3 + CD4 - CD8 - T-linfocitos (doble negativo) en la sangre periférica y un defecto de la apoptosis [defecto del gen Fas.(CD95) - TNFRSF6 (10T24.1),goma cASPAZA-10,FAS LIGAND - FASL (1T23)].
Datos de laboratorio.El contenido de CD3 + CD4 - CD8 - T-Lymphocytes en sangre periférica o en tejidos linfoides es más del 1%. El nivel IgG, IgA y IgM puede ser normal, elevado o incluso reducido. Con la edad, la hipergammaglobulinemia se sustituye por una baja concentración de inmunoglobulinas séricas, hasta Aghamglobulinmia. Revelar autoantibodes a glóbulos rojos, plaquetas, neutrófilas, músculos lisos, para factorizar VIII; Los autoanticuerpos antinucleares y anti-fosfolípidos, así como el factor reumatoide, etc. se caracteriza por la linfocitosis.
Cuadro clinico.Todos los pacientes han aumentado el hígado, los ganglios linfáticos (en los primeros 5 años de vida) y el bazo. La linfoproliferación no está acompañada por un aumento de la temperatura y la sudoración nocturna. Debut reacciones autoinmunespuede que no coincida con la linfoproliferación y viene más tarde. Con la edad, la severidad de las reacciones autoinmunes está aumentando. Las reacciones autoinmunes contra las células sanguíneas (anemia hemolítica, trombocitopenia, neutropenia) se están desarrollando con mayor frecuencia que otros órganos. El riesgo de desarrollar neoplasias malignas (t-y b-linfoma, linfoma de Berkitt, linfoma atípico, linfogneumatosis, etc.).
Tratamiento.. Agentes quimioterapéuticos (ciclofosfamida, azatioprina, metotrexato, clorambucil). . Glucocorticoides. . Splenectomía con hypranismo pronunciado y hemocitopenia. . En casos severos, el trasplante de médula ósea es posible.
Síndrome de hiperimmunoglobulinemia E.
El síndrome de Hyper-IgE se caracteriza por un aumento significativo en el nivel de IgE sérico, los abscesos repetidos de la piel y la fibra subcutánea de etiología estifilocócica, neumonía con la formación de neumáticas, anomalías de la estructura de esqueleto facial, ATD. La naturaleza genética molecular del síndrome de Hyper-IGN aún no se ha establecido. En algunos casos, autosomalizado, en otros, se revela la herencia autosómica-recesiva. Se supone que los defectos afectan las moléculas de señalización de los receptores de citoquinas (en la forma autosómica dominante de este síndrome, ya se han revelado mutaciones en Stat3)y, posiblemente asociado con una violación del funcionamiento de la subpoblación de las células TH17. Otro gen responsable de la formación del síndrome de hiper-ign, localizado en el cromosoma 4. (4Q).
Datos de laboratorio.Revelar una variedad de trastornos inmunológicos: aumentar el nivel de IgE en suero, quimiotaxis deteriorada de los neutrófilos, defectos para la formación de anticuerpos; Reduciendo la reacción de la GZT a candidina, difteria y anatoxinas del tétanos; Debilitamiento de la actividad proliferativa de las células T en respuesta a Candida.y el tétanos anatoksin mientras mantiene una respuesta a los mitógenos. Eosinofilia en sangre periférica y abscesos de la piel líquida. El número de células T y B es normal.
Cuadro clinico.Eczema del curso de medio paso a una edad temprana. Características características de la cara (puente más ancho, nariz de humo ancho, asimetría del esqueleto facial, sobresaliendo frente, ojos profundamente plantados, paladar alto). Anomalías para el desarrollo de un esqueleto, escoliosis, mayor movilidad de articulaciones, tendencia a las fracturas de los huesos después de lesiones menores, violación del cambio de dientes. Surgen los abscesos de la piel, las fibras subcutáneas y los ganglios linfáticos surgen. La neumonía se está desarrollando a una edad avanzada (patógenos más frecuentes. S. aureus.y H. IN-
fluenzae),en el 77% de los casos, se forma Pneumocele, se unió una infección debido a P. aeruginosa.y A. fumigatus.La neumonía puede fugarse sin aumentar la temperatura. La candidiasis crónica de las membranas mucosas y las uñas se está desarrollando en el 83% de los casos.
Tratamiento.. Larga (para el propósito de la prevención, la terapia antibacteriana y antifúngica). . Para tratar la dermatitis, los agentes tópicos se utilizan en casos pesados, dosis bajas de ciclosporina A .. El trasplante de médula ósea es ineficaz.
Síndromes cromosomas de desglose
Para síndromes con inestabilidad cromosómica: attokiatelEngitasia[DEFECTO DEL GENE DE LA TOPOISOMERA DE ADN ATM (11T22),pI. síndrome Nijimegen[Defecto del gen Nibrino Nbs1(8T21)] - caracterizado por una mayor frecuencia de tumores malignos, inestabilidad cromosómica espontánea y desgloses cromosómicos. Ambas proteínas participan en las reparaciones de las brechas de la litera de ADN y que regulan el ciclo celular. Normalmente, las roturas de unión al ADN se producen en el proceso V (D) J de recombinación de genes de inmunoglobulina y TCR, cambiando las clases de inmunoglobulina, durante el reticulante y durante la meyosis. Se producen procesos similares en la maduración de las neuronas cerebrales. Los defectos de reparación de ADN en la ataxia-teleangecto y el síndrome de Niimgen causan dichas manifestaciones clínicas, como la síntesis de inmunoglobulina deteriorada, la función de los órganos genitales y el sistema nervioso.
Ataxia telangitasia
Este síndrome (frecuencia de 1: 300 mil recién nacidos) con un fenotipo muy heterogéneo describió el médico francés D. Louis-Bar. Los síntomas de la ataxia se pueden encontrar en el niño ya a la edad de 2 a 4 meses. La ataxia se debe a la degeneración progresiva de las células Purkinier en el cerebelo. Las teleangicas en la piel de la nariz, los lavabos de oído, en la conjuntiva aparecen un poco más tarde, en 3-6 años. A menudo, en la piel, hay manchas como "café con leche". Tymus hipoplasia, ganglios linfáticos, bazo, almendras son característicos. La inmunodeficiencia se muestra mediante una disminución (a menudo desequilibrada) IgA, IgE, IgG2, IgG4. El 80% de los pacientes se desarrollan.
síntomas clínicos infecciosos apropiados. Se reduce el número y la actividad funcional de las células T (principalmente células T CD4 + T). El número total de linfocitos T en la mayoría de los pacientes es normal. Extremadamente alto (200 veces más alto que en la población general) la frecuencia de las neoplasias (principalmente linfoma y carcinomas), a menudo lleva a la muerte en 10-12 años. Tratamientosintomático.
Síndrome Nijimegen
Síndrome Nijmegen (por el nombre de la ciudad en Holanda, donde se describió por primera vez la enfermedad) manifestada por microcefalus, un esqueleto facial con discapacidad específico (frente biselada, sobresaliendo parte media de la cara, nariz larga, hipoplasia de la mandíbula inferior, el corte de ojos mongolaid, los epicanti, grandes oídos), el retraso del desarrollo físico, la presencia de hoteles "café con leche" en la piel; Klianodactilia y Syltons, disgenesia de ovario y otros. La mayoría de los niños sufren de bacterias recurrentes y crónicas. enfermedades infecciosastracto respiratorio, órganos lora y sistema urinario. En el 50% de los casos, las neoplasias malignas se están desarrollando, principalmente en linfomas celulares. Se revelan varias formas de disbammaglobulinemia, una disminución en células T CD4 +.
Tratamiento.. Terapia sintomática de trastornos neurológicos. . Terapia de reemplazo con inmunoglobulina intravenosa. . De acuerdo con las indicaciones, se usa antibacterianos, antivirales, terapia antifúngica. . En el tratamiento de neoplasias malignas, se tienen en cuenta el aumento de la sensibilidad a los productos de radiación y quimioterapia.
Síndrome de Viscott-Aldrich
Síndrome de Viscott-Aldrich [Defecto Gene WASP (xp11.23p11.22),אּ; también ρ y ʀ] gen Avispa.(de Síndrome de Wiskott-Aldrich)expresado en telas de linfocitos, bazo y tymocitos. Las mutaciones de este gen están asociadas con la expresión anormal en los neutrófilos y las moléculas CD43 (CD4 y CD8) (LIGAND para ICAM-1, realizan una función antiadhesiva).
Datos de laboratorio.La trombocitopenia (menos del 10% de la norma) se debe al aumento de la destrucción de las células.
Lesas de plaquetas que en personas sanas. El nivel de IgM en suero sanguíneo se reduce en el nivel normal de IgG y un aumento en el contenido de IgA y IgE. Se reducen los títulos de isohemaglutininas, se reduce la formación de anticuerpos contra los antígenos de polisacárido de los palitos de neumocócicos, estreptocócicos, intestinales, salmonela, así como anticuerpos antivirales. A una edad temprana, como regla general, el número de linfocitos es normal, después de 6 años detectan linfoprio (menos de 1x10 9 / L), una disminución en células T CD3 + y CD4 + en el nivel normal de células V- y NK . La eosinofilia y el desarrollo de la anemia postghemorrágica son posibles. La respuesta proliferativa de las células T en los mitógenos y antígenos se reduce, GZT se debilita. En el bazo, las estructuras normales de los centros germinativos y las zonas de células T no se detectan.
Cuadro clinico.La enfermedad se caracteriza por signos de tríadas: trombocitopenia, eccema y enfermedades infecciosas recurrentes.El síndrome de hemorrágico se manifiesta temprano, ya durante el período de recién nacida (erupción fetecera, cheefalohematomas, sangrado de heridas umbilicales, sangrado intestinal). El eccema se manifiesta de una edad temprana en el 80% de los pacientes. Los signos de inmunodeficiencia están aumentando con la edad: enfermedades infecciosas bacterianas de los órganos ENT, sistema respiratorio, órganos digestivos, piel; Infección herpética común o generalizada. (Herpes Simpley Varicela zoster),citomegalovirus, así como fúngico (candidiasis de membranas mucosas), menos a menudo enfermedades infecciosas oportunistas. En el 70% de los pacientes identifican enfermedades autoinmunes (anemia hemolítica, neutropenia, artritis, vasculitis de la piel, colitis ulcerosa no específica, vasculitis cerebral, glomerulonefritis, trombocitopenia autoinmune). Los pacientes mayores de 5 años aumentaron la frecuencia. neoplasmas malignos(en su mayoría tumor de tejido lempoide).
Tratamiento.. Trasplante de la médula ósea alogénica o las células madre (el éxito de la operación alcanza el 90% cuando se usa un injerto de un donante histocompatible y el 50%, con trasplante haploidental). . Terapia de reemplazo con inmunoglobulina intravenosa. . Propósito preventivo de drogas antibacterianas, antifúngicas y antivirales. . Para reducir el síndrome de hemorrágico, se lleva a cabo la esplenectomía. . En las complicaciones autoinmunes, se prescriben la terapia inmunosupresora.
Defectos de fagocitosis
Enfermedad granular crónica
La enfermedad de Granuner crónica se caracteriza por una violación de la actividad funcional de los fagocitos (la formación de formas activas de los radicales de oxígeno, el asesinato intracelular y la fragmentación de los patógenos fagocytados), las enfermedades infecciosas bacterias y infecciosas permanentes y el desarrollo de la inflamación granular. Enfermedad granular crónica en personas con diferentes defectos genéticos [en el 65% de los casos - versión vinculada X de la enfermedad: gen gP91-Phox (XP21.1),אּ; En el 35% de los casos - autosomal-recesivo: gen f47-PHOX (7T11.23),ρ; gene p67-PHOX (1T25),ρ; gene p22-PHOX (16Q24),p], lo que lleva a violaciones en el sistema NADF-Oxidasa. Con la muerte de los neutrófilos de corta duración (varias horas), las bacterias no unidas "Fugas" en el enfoque de la inflamación. Macrófagos: las células de larga duración, y sus predecesores (monocitos) migran al hogar en mayores cantidades (lo que conduce a la formación gránulos)los microorganismos fagocíticos, pero no pueden matarlos.
Datos de laboratorio.Los valores normales de las inmunoglobulinas de suero y las subpoblaciones de los linfocitos son característicos. La formación de radicales peroxidantes por neutrófilos, se estima en las pruebas (la quimioluminiscencia dependiente de luminicación o la restauración del azul tetrazolio), se reduce o ausente. En el contexto de las enfermedades infecciosas, la leucocitosis, la neutrófilosis, un aumento en la ESR, la anemia, la hipergammaginemia.
Cuadro clinico.La enfermedad en la mayoría de los casos se manifiesta en el primer año de vida. síndrome infeccioso(Infecciones internas y patógenos extracelulares) y la formación de un gránulo. Los más típicos: daño pulmonar (neumonía recurrente, lesión de los ganglios linfáticos de tostado, abscesos pulmonares, pleurisitas purulentas), tracto digestivo, abscesos de la piel y linfadenitas. Los patógenos más frecuentes - Microorganismos positivos de catalasa: S. Aureus, Aspergillus spp.,bacterias intestinales gramnegativas (E. coli, Salmonella spp., Serratia Marcescens),menos probable - Burkholderia Cepacia.y NOCARDIA FARCINICA.El desarrollo de abscesos hepáticos y subadiáfragmales, osteomielitis, abscesos paráneos, sepsis son característicos.
La complicación infecciosa que amenaza la vida más grave es la aspergilosis, que puede ocurrir en forma de lesión difusa de pulmones y otros órganos (tejido graso, cerebro, huesos, articulaciones, endocardio). En pacientes con enfermedad granular crónica después de la vacunación, BCG a menudo desarrolla infecciones asociadas a la vacuna que involucran los ganglios linfáticos regionales. El daño micobacteriano en pacientes con enfermedad granular crónica puede tener la localización ligera y extrema y un flujo prolongado. Para los pacientes con enfermedad granular crónica, el retraso en el desarrollo físico es característico.
Tratamiento.. Terapia antimicrobiana: uso constante profiláctico de co-trimoxazol y fármacos antifúngicos (itraconazol et al.); En caso de complicaciones infecciosas, se realiza la terapia antibacteriana combinada por pariente (2-3 antibióticos bactericidas penetrantes intracelulares) en combinación con la terapia antifúngica. Al desarrollar la aspergilosis, se muestra un uso a largo plazo de la anfotericina en o Kaspofung. En la infección micobacteriana, se utiliza una combinación de terapia específica a largo plazo con medicamentos anti-tuberculosis con una amplia gama de antibióticos de acción. . El tratamiento quirúrgico a menudo está acompañado por la supresión de la herida postoperatoria y la formación de nuevos focos. Es posible un drenaje puntual de un absceso bajo el control del ultrasonido. . Para el tratamiento de complicaciones infecciosas pesadas en la ineficacia de la terapia antibacteriana, es posible el uso de masa granulocítica, altas dosis de IFNU y G-CSF. . El trasplante de la médula ósea o el trasplante de los células sanguíneos del cordón de la sangre del hermano compatible pueden tener éxito a una edad temprana cuando minimiza el riesgo de muerte contra las complicaciones infecciosas y la reacción "trasplante contra el propietario".
Adhect adhect leucocyte
Hasta la fecha, se describen 3 defectos de adhesión de leucocitos. Todos tienen
un tipo de herencia autosómico-recesivo se caracteriza por enfermedades infecciosas y fúngicas crónicas y crónicas. El tipo I se caracteriza por la ausencia o disminución de la expresión de CD11 / CD18 en los leucocitos, la penetración de los neutrófilos, la leucocitosis (más de 25x10 9), más adelante la deposición del residuo del cordón y el desarrollo de la omnipalitis, la mala curación de Heridas, la ausencia de la formación de pus en la penetración del camino del patógeno en el cuerpo.
Tratamiento.. Terapia antibacteriana: episodios infecciosos y profilácticos. . En caso de flujo pesado, se prescribe el trasplante de médula ósea del donante compatible con HLA.
Defectos del sistema de complemento.
Enfermedades con el déficit de componentes del complemento.
Las manifestaciones de los defectos genéales de los componentes individuales del sistema de complemento se dan en la tabla. 11-2.
JSC saludable.Las enfermedades causadas por un déficit de complementos de complemento rara vez se detectan, ya que el estado homocigótico de alelos autosómicos es necesario para su manifestación. Hay una única exclusión asociada con C1inh (inhibidor C1-esterasa): mutación del gen C1inh,el inhibidor que conduce a la deficiencia en el estado heterocigoto se manifiesta por un fenotipo, conocido como la AO hereditaria (ver más Capítulo 13, Angio-Movimientos).
Enfermedades de los complejos inmunes.La insuficiencia de C1-C4 se manifiesta por el desarrollo de enfermedades de los complejos inmunes: vasculitas sistémicas y daños causados \u200b\u200ben el riñón, que se generaliza denominado síndrome de lupus rojo sistémico (SC).
Infecciones pyiogénicas.La deficiencia de C3 (también factores H y I) se asocia con una mayor susceptibilidad a las infecciones por pirógenos. El déficit de los componentes involucrados en la trayectoria alternativa de la activación del complemento, así como la deficiencia de los componentes C5-C8 asociados con una mayor susceptibilidad a la infección causada por Neisseria spp.La deficiencia de C9 suele ser clínicamente asintmésica.
Tabla 11-2.Manifestaciones clínicas de defectos de componentes individuales del sistema de complemento.
LESTEN DE DEFICIENCIA DE DEBENCIÓN MANNOSA
Conexión de la deficiencia de liquen de manosa (Otro Nombre: MannanAbying Lectin - MSL) se debe a un defecto genético MBL.(Diferentes puntos de mutaciones y eliminaciones en el gen. MBL.revelar del 17% de la carrera de la gente europea). Con los defectos del gen, la activación de las proteasas, dividiendo los componentes del complemento C2 y C4, y se altera la activación del sistema de complemento en la vía de lectina. Clínicamenteesta patología se manifiesta por síndrome infeccioso.
Datos de laboratorio.El análisis de las subpoblaciones de los linfocitos, los leucocitos, así como los isotipos de inmunoglobulina no muestran desviaciones significativas adecuadas a los síntomas clínicos. No hay MSL en suero.
Tratamiento.Esta enfermedad no es una inmunodeficiencia clásica. En consecuencia, los medios inmunotrópicos inmunocorrectivos están contraindicados. La MSL recombinante se puede usar como una preparación farmacológica para la terapia de sustitución etiopatogenética de los pacientes con este defecto hereditario. Actualmente, este medicamento pasa los ensayos clínicos.
Hipogammaglobulinemia transitoria en niños
La hipogammaglobulinemia transitoria en niños está asociada con una característica fisiológica de la formación gradual del sistema de inmunoglobulina. La maduración de la formación de anticuerpos IgM y IgA es la mayoría de los "retrasos". En niños sanos, el contenido de la IgG materna disminuye gradualmente y después de medio año aumenta el desarrollo de sus propios anticuerpos IgG. Sin embargo, en algunos niños, se retrasa el aumento en el nivel de inmunoglobulinas. Tales niños pueden sufrir enfermedades infecciosas bacterianas recurrentes. En estos casos, no debe recurrir a las infusiones de las drogas de las inmunoglobulinas de los donantes (introducción de inmunoglobulina intravenosa).
Deficiencia selectiva de inmunoglobulina a
Inmunoglobulina A (SD IGA - Deficiencia selectiva de IgA)desarrollado como resultado de un defecto genético. tNFRSF13B.
o p). La deficiencia de IGA en presencia de inmunoglobulinas de otras clases es la inmunodeficiencia más frecuente detectada en una población común con una frecuencia de 1: 500-1500 personas (en pacientes que sufren alergias, más a menudo). Insuficiencia selectiva de IGA, es decir, Contribuyendo en una escasez de una de las subclases (30% de los casos) y completa (70% de los casos). La deficiencia de la subclase IgA2 conduce a una imagen clínica más pronunciada que el déficit de la subclase IgA1. Las combinaciones de deficiencia de IgA con otras violaciones son posibles: con defectos de biosíntesis de IgG y anomalías de linfocitos T. La gran mayoría de las personas con selectividad.
la deficiencia de la IGA es prácticamente saludable. Para niños menores de 2 años, la deficiencia de IgA es un estado fisiológico.
Revelar la reducción de la concentración sérica de IgA a<5 мг/дл у детей старше 4 лет; IgG и IgM в норме, количество и соотношение субпопуляций лимфоцитов и их функциональная активность могут быть в норме.
Cuadro clinico.Con una deficiencia de IgA, se pueden desarrollar 3 grupos de síndromes patológicos: infecciosos, autoinmunes y alérgicos. Los pacientes con deficiencia de IgA están predispuestos a enfermedades infecciosas recurrentes del tracto respiratorio superior y los órganos digestivos. Los más frecuentes y gravemente fluyen son una variedad de enfermedades autoinmunes (artritis reumatoide, spondilitis anquilosante, síndrome de granos, vasculitis con una lesión de vasos cerebrales, tiroiditis autoinmune, LES, glomerulonefritis, anemia hemolítica, diabetes tipo I, vitiligo, etc.). La frecuencia de Celiachood excede tales en niños con IgA normal 10 veces. Las manifestaciones alérgicas más detectadas: la intolerancia a la proteína de la leche de vaca, la dermatitis atópica (ATD), el asma bronquial.
Tratamiento.Los casos de flujo asintomático no requieren ningún tratamiento especial; En presencia de manifestaciones clínicas de enfermedades infecciosas, autoinmunes y alérgicas, el tratamiento se realiza de acuerdo con las normas.
La terapia de reemplazo de las inmunoglobulinas de los donantes no se muestra en selectiva, ni con una deficiencia completa de la IGA, ya que la probabilidad de la formación de anticuerpos anti-etióticos a la IGA y el desarrollo de complicaciones de transfusión causadas por ellos es alta.
Agamaglobulinemia con deficiencia de células B
Aghamaglobulinemia agarrada X (enfermedad de Bruton)es el 90% de todos los casos de Aghamaglobulinemia. Los niños están enfermos, hijos (אּ, ρ) de portadores de un gen defectuoso bTK (XQ21.3-Q22),proteinthyrosinkinasa proteinthyrosinkinasa B-Lymphocyte BTK (Bruton "s tirosina quinasa- Bruton tirosina quinasa). Como resultado del defecto, una violación de las trayectorias de señalización intracelular, la recombinación de las cadenas de inmunoglobulina pesadas, difunto
recuperación de células BR B en los linfocitos B. En el 10% de los pacientes con agammaglobulinemia con deficiencia en células, se hereda la autosomongénesis. 6 Los defectos genéticos se describen actualmente, incluidas las moléculas del receptor de células pre-B, las células B del adaptador citoplásmico (BLNK) y el gen Rico en leucina que contiene 8 (LRRC8).
Datos de laboratorio.No hay linfocitos B periféricos. En la médula ósea hay células pre-B con una cadena μ en el citoplasma. El número de linfocitos T y las pruebas funcionales en los linfocitos T pueden ser normales. IgM y IgA en la sangre no pueden ser revelados; IgG puede estar presente, pero en pequeñas cantidades (0.4-1.0 g / l). No hay anticuerpos contra los antígenos de los grupos sanguíneos y los antígenos de la vacuna (tétanos, toxinas difterias, etc.). La neutropenia puede desarrollarse. Estudio histológico de la tela linfoide: no hay centros germinativos (germinales) y células plasmáticas en los folículos linfoides.
Cuadro clinico.Si el historial familiar es desconocido, el diagnóstico se vuelve obvio en promedio durante 3,5 años. La enfermedad se caracteriza por un tejido linfoide de hipoplasia, infecciones purulentas que fluyen pesadamente, enfermedades infecciosas de la parte superior (sinusitarios, otitis) y menor (bronquitis, neumonía) del tracto respiratorio; Gastroenteritis, piodermia, artritis séptica (bacteriana o clamazdiosa), septicemia, meningitis, encefalitis, osteomielitis. Como agentes causales de enfermedades respiratorias, la mayoría de las veces se realiza. Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus,bacterias intestinales de la diarrea o Giardia Giardia Lamblia.Además, los pacientes con Aghamaglobulinemia son susceptibles a enfermedades infecciosas causadas por micoplasmas y ureasmos que causan el desarrollo de neumonias crónicas, artritis purulenta, cistita y absceso subcutáneo. Desde virus, virus neurotrópicos típicos ECHO-19 y cokes, causando tanto la encefalitis pesada y crónica y la encefalomielitis. Las manifestaciones de las infecciones de enterovirus pueden ser un síndrome de dermatomía, una ataxia, dolores de cabeza, violaciones de comportamiento. En pacientes con niños en inmunización, un poliocresino vivo, como regla general, se detecta una liberación a largo plazo del virus de la polio a través de las membranas mucosas, y con la virulencia restaurada y creciente (es decir, en el colector de niños.
somos un peligro real de infección de niños sanos con polio, como resultado del contacto con un niño inmunodeficiente vacunado). Los trastornos autoinmunes durante la agammoglobulinemia pueden representarse por artritis reumatoide, síndrome de esclerodermo, escleroderm, colitis ulcerosa no específica, diabetes tipo I (debido a la predominio de la respuesta inmune TH1).
Inspección física.Preste atención al retraso en el desarrollo físico, en la forma de los dedos (dedos en forma de palos de tambor), cambios en la forma del tórax, característicos de las enfermedades del tracto respiratorio inferior, la hipoplasia de los ganglios linfáticos y las almendras.
Tratamiento.
Terapia de reemplazo: las fármacos de inmunoglobulina intravenosa se introducen cada 3-4 semanas para la vida. Las dosis de inmunoglobulinas se seleccionan para crear en el suero del paciente su concentración, superponiendo al límite inferior de la norma de edad.
Discutir la posibilidad de terapia génica - gen BTK.se cloneó, sin embargo, su hiperexpresión está asociada con una transformación maligna del tejido hematopoyético.
En el caso de la neutropenia persistente, se utilizan factores de crecimiento. Con la aparición de signos de patología autoinmune, es posible designar medicamentos de anticuerpos monoclonales (infliximab, etc.).
Fracaso inmune variable general
La falla inmune variable común (OVIN) es un grupo de síndromes caracterizados por un defecto para la síntesis de anticuerpos y la inmunidad celular. El criterio de diagnóstico confiable de AVIN es una reducción significativa en el contenido de las inmunoglobulinas de dos o tres isotipos principales en personas de ambos sexos en combinación con uno de los siguientes signos:
Debut de la enfermedad mayor de 2 años;
Falta de isohemaglutininas y / o baja respuesta de vacunación;
Una exclusión de otras razones de Aghamglobulinemia.
En algunos pacientes, la causa del desarrollo de AVIN es la mutación de los genes que codifican moléculas involucradas en los procesos de maduración y supervivencia de las células B: BAFF-R (Receptor de factor de activación de células B)BLIMP-1. (Proteína de maduración inducida por linfocitos B)y iCOS. (Costimulador inducible).Existe una violación de la capacidad B-Lymphocyte para diferenciarse en células plasmáticas, se están desarrollando defectos de anticuerpos, se está desarrollando disfunción en T linfocitos, se observa una mayor tendencia a las enfermedades infecciosas. El síndrome puede manifestarse en la primera infancia, en la adolescencia o en los jóvenes.
Datos de laboratorio.Los niveles de IgG e IgA se reducen significativamente (aproximadamente el 50% de los pacientes) y IgM (hasta la discusión). El número de linfocitos B en la sangre corresponde a la norma o reducido. El número de linfocitos T en la mayoría de los pacientes es normal. En pacientes graves, es posible desarrollar linfopenia (menos de 1500x103 células en 1 litro de sangre). El número de células NK se reduce. La producción de anticuerpos específicos en respuesta a la inmunización se reduce o ausente. La proliferación de linfocitos y la formación de IL-2 bajo la acción de mitógeno y antígenos se violan significativamente.
Cuadro clinico.Reclutar enfermedades infecciosas bacterianas recurrentes con localización predominantemente en el tracto respiratorio y los senos incompletos. En el momento del diagnóstico de la infección respiratoria, el tracto respiratorio puede progresar a la bronquiectasia y las lesiones derramadas de tejido claro. Es posible un daño infeccioso a los órganos digestivos, manifestados por la diarrea, el vapor y la malabsorción (y, en consecuencia, la pérdida del peso corporal). A menudo identifica infecciones causadas por Giardia Lamblia, Pneumocystis Cariniio virus familiares Herpetoviridae.Los pacientes de AVIV son propensos al desarrollo de la artritis purulenta causada por micoplasmas y ureapasmos. Las manifestaciones de las infecciones por enteravirus pueden ser la encefalomielitis, los síndromes polémicos y similares a la dermatomía, lesiones de la piel y las membranas mucosas. Autoinmunelas enfermedades ocurren duras y pueden determinar el pronóstico de OVIN. A veces, las primeras manifestaciones clínicas de AVIVE son la artritis, la colitis ulcerosa no específica y la enfermedad de Crohn, la colangitis esclerosante, la malabsorción, el LES, la nefritis, la miositis, el daño autoinmune a los pulmones en forma de neumonita intersticial linfoide, neutropenia,
púrpura trombocitopénica, anemia hemolítica, anemia perniciosa, alopecia total, vasculitis retina, fotosensibilización. En pacientes, el ovino tiene una frecuencia significativamente mayor. neoplasmas malignos(En el 15% de los casos), el gránulo similar a los sarcoidos y la linfoproliferación no maligna. Tratamiento.
Quimioterapia antobacteriana.
Terapia de reemplazo: las fármacos de inmunoglobulina intravenosa se introducen cada 3-4 semanas para la vida.
En las complicaciones autoinmunes, la terapia inmunosupresora (glucocorticoides, la azatioprina, la ciclosporina A) y es posible prescribir preparaciones de anticuerpos monoclonales (infliximab, etc.).
Síndromes de Hyper-IgM
Los síndromes de Hyper-IgM son enfermedades bastante raras caracterizadas por una disminución pronunciada o ausencia completa de IgG, IgA y una concentración de IgM en suero normal o elevada. Esto es causado por la incapacidad de los linfocitos B para cambiar las clases de inmunoglobulina y la hipermutagénesis de dominios variables. Hasta la fecha, se han identificado 6 defectos genéticos que llevan al desarrollo de Hyper-IGMSPROM.
. TIPO 1 (HIGM 1).Deficiencia de LIGAND DE LIGAND DE LIGNADA X (70% de los síndromes de Hyper-IgM), que conduce a la incapacidad de las células T para interactuar de manera efectiva en los linfocitos.
. Tipo 2 (HIGM 2).Autosomal-recesivo, asociado con un defecto de ayuda, la activación inducida por la citidina deamindaasa (gen AICIDA, 12P13)- Enzima involucrada en las clases de conmutación de inmunoglobulinas e hipermutagénesis.
. Tipo 3 (Higm 3).Autosomal recesivo, asociado con la mutación del gen de la molécula CD40. Al mismo tiempo, las células B en sí mismas no pueden interactuar de manera efectiva con los linfocitos T. Las manifestaciones fenotípicas son similares a las de tipo 1.
. Tipo 4 (HIGM 4).Autosómica recesiva; En algunos casos, surgen mutaciones. de Novo.Sonaba con un ung - uracil-dna-glicosyase - enzima, que también participa
en las clases de conmutación de inmunoglobulinas, pero después de la acción de ayuda. En este caso, la hipermutagénesis no afectará y el síndrome fluye con menor severidad.
. Tipo 5 (Higm 5).Defecto solo en las clases de conmutación, la hipermutagénesis no afectará. La mutación causal aún no se ha detectado, pero, obviamente, el defecto en la enzima actuando después de
. TIPO 6 (HIGM-ED).X-alojado, asociado con una displasia ectodérmica de descarga, causada por una deficiencia de NEMO (modulador NF-KB), lo que lleva a una violación de transmisión de señal con CD40.
Síndrome de Hyper-IgM con Transferencia Xrevelar más a menudo. Desarrollado cuando un defecto de genes de codificación CD40L (CD154, el gen se encuentra en Xq26-q27.2)- Ligand para CD40. La insuficiencia de la expresión de los linfocitos T CD40L conduce a la imposibilidad de cambiar las clases de inmunoglobulina en los linfocitos B con IgM a otros isotipos, así como a una violación de la formación de células B de memoria, repertorio de células T y TH1. Respuesta celular dirigida contra microorganismos intracelulares. Los niños están enfermos
Datos de laboratorio.IgG, IgA, IgE no se puede determinar ni detectar en cantidades muy pequeñas. El nivel IGM es normal (en el 50% de los casos) o elevado, a menudo significativamente. La cantidad de células T y B es normal; Reducción de la respuesta proliferativa de las células T inducidas por los antígenos. IgM es policlonal, a veces monoclonal. Se detecta antena terapia del isotipo IgM (anti-aleatorio, antitrombocito, anti-aleatorio, anticuerpos a los antígenos de tejido muscular liso). En tela linfoide, no hay centros germinativos, pero hay células plasmáticas.
Cuadro clinico.Las primeras manifestaciones surgen en la infancia infantil y temprana. Característico repetido infecciónvarias localizaciones (principalmente tracto respiratorio), incluyendo oportunistas (causadas Pneumocystis carinii).También característica de los virus (citomegalovirus y adenovirus), Criptococcus neoformans,mycoplasmas y Mycobacteriums. La infección criptosporidial puede causar diarrea aguda y crónica (desarrollándose en el 50% de los pacientes) y la colangitis esclerosante. Anemia, neutropenia, ulceración de la mucosa oral, gingivitis, ulcerativa.
abejas del esófago, varios departamentos intestinales, colitis ulcerosa no específica. Revelar la predisposición a K. trastornos autoinmunes(Artritis seronegativa, glomerulonefritis, etc.) y neoplasias malignas (principalmente tejido linfoide, hígado y tracto biliar). Linfadenopatía, hepato y esplenomegalia pueden desarrollarse. Tratamiento
Terapia de reemplazo regular con inmunoglobulina intravenosa.
Quimioterapia antobacteriana. Para la prevención y el tratamiento de los neumonios neumónicos, se utilizan co-trimoxazol [sulfametoxazol + trimethoprim] y pentamidina.
Para evitar la lesión del hígado y el tracto biliar, solo se debe usar agua hervida o filtrada, examine regularmente (examen de ultrasonido, biopsia hepática de acuerdo con las indicaciones).
En el tratamiento de la neutropenia y las ulceraciones de la cavidad oral, se utilizan glucocorticoides y preparaciones de un factor de colonización de granulocitos.
En el desarrollo de complicaciones autoinmunes, se prescriben terapia inmunosupresora (glucocorticoides, azatioprina, ciclosporina A), así como medicamentos basados \u200b\u200ben anticuerpos monoclonales.
El método óptimo de tratamiento es el trasplante de médula ósea de los donantes compatibles con HLA (el nivel de supervivencia es del 68%, es mejor conducirse menor de 8 años).
Esta es la inmunodeficiencia primaria más común. Ocurre con una frecuencia de 1/300 - 1/700. Primero fue ...
Descrito por J. Heremans en 1960.
En la mayoría de los casos, no se observa una imagen clara de la herencia de la enfermedad, en casos más raros de la pérdida familiar de la IGA, la herencia autosómica-recesiva ocurre con más frecuencia que los dominantes. La deficiencia de IGA a veces se asocia con las anomalías del cromosoma 18.
La IGA difiere de otras inmunoglobulinas en el contenido de carbohidratos y ácidos sialicos y la capacidad de formar dímeros, trímeros e incluso tetramers. Distinguir 2 opciones para las moléculas de IgA:
- SERUM IGA - SIEMPRE MONOMER;
- Secretor IgA - Forma di-, tres y tetramers, antes de convertirse en una parte integral de los secretos (saliva, desgarro, leche, secretos de tractos digestivos, respiratorios y urinarios).
Una cadena especial de proteínas está participando en la educación de la Cadena Secretaria IgA - J. En consecuencia, este es un sistema de proteínas entero. A partir de aquí - Formas de patología IGA:
- El fallo general de la IGA está asociado con las anomalías de la síntesis del monómero IgA. Como resultado, se reducen el contenido y el suero y la IGA secretor. La protección local y general está perturbada;
- Defecto para la formación de moléculas secretoras - SIGA. La razón puede ser la ausencia de una cadena J, que conduce a una violación de la inmunidad local;
- Violación preferencial de la síntesis de la IGA sérica, cuando solo se asigna las formas de IGA secretor, y prácticamente no se secretan los monómeros de IgA en suero.
Los síntomas clínicos dependen de grados de deficiencia de IgA Y pueden manifestarse en forma de patología de órganos y sistemas individuales. La mayoría de las veces hay daños infecciosos a las membranas mucosas.
Desde la cabeza del tracto gastrointestinal es la gastritis crónica, la colitis ulcerativa y hemorrágica, Ileit, estomatitis. Desde el lado de las autoridades respiratorias, la rinitis, los sintitis, la bronquitis, que recibe rápidamente el carácter crónico. A menudo hay bronconeumonía crónica con un resultado en bronquiectasa y abscesos pulmonares. También se detectó la conexión de un contenido reducido de la IGA secretor con una mayor morbilidad de las alergias atópicas y con el desarrollo de enfermedades autoinmunes (lupus rojo sistémico, artritis reumatoide, tiroiditis autoinmune).
En las personas con un déficit selectivo de la IgA predominantemente del monómero sérico, ninguna manifestación clínica de la inmunodeficiencia ocurre con mayor frecuencia.
Investigación de laboratorio:se muestra que el número total de linfocitos T y B en la sangre periférica en tales pacientes está dentro del rango normal. Los niveles de IGA sérico y / o en secreto se reducen considerablemente, mientras que el contenido de IgM e IgG está dentro del rango normal. En pacientes individuales (con manifestaciones de alergias atópicas), el nivel de IgE puede ser elevado.
Tratamiento: sintomático. Es necesario tener cuidado al usar la terapia de sustitución con inmunoglobulinas, ya que cuando la administración repetida existe el riesgo de un choque anafiláctico debido a la posibilidad de aumentar los productos anti-inmunoglobulina de las clases IgG y IgM.
Pronóstico: El curso de la enfermedad es principalmente benigno.
Criterio de diagnóstico - Reducido en pacientes mayores de 4 años de niveles de IgA sérico inferior a 0,07 g / l con contenido de IgG normal y IgM y la exclusión de otras causas de hipogammaglobulinemia. Diagnósticamente significativo:
Una reducción aislada en los niveles séricos de la IGA (menos de 0.05 g / L) en el nivel normal de otros isotipos de inmunoglobulina en niños de más de 1 año, sin Igal e IgA2. Los niveles de IgM e IgG son normales. Sin embargo, algunos pacientes identifican la deficiencia de IgG2;
- Si el nivel IGA está en el rango de 0.05 g / l a 0.2 g / L, entonces se diagnostica la insuficiencia parcial de la IGA; el número normal de linfocitos T y sus subclases;
- generalmente el número normal de linfocitos en los linfocitos (CD19 \\ CD20);
- Normal NK Cell (CD16 CD56).
En pacientes con deficiencia de IgA.Especialmente en ausencia de IgA secretor, es necesario explorar el nivel de subclase IgA. En algunos pacientes, la insuficiencia selectiva de la IGA puede progresar con el desarrollo en el futuro de AVIV. Se necesita un largo monitoreo regular del contenido de las inmunoglobulinas (incluidos los pacientes asintomáticos).
Determinación de Autoantile (antinuclear, antihiidroid, etc.).
Con comida intolerancia O malabsorción necesita alergia y definición de anticuerpos contra leche y anticuerpos IgG anti-adversos.
Tratamiento. No se requiere pacientes con deficiencia selectiva asintomática del tratamiento constante de IgA. Los pacientes con manifestaciones de enfermedades infecciosas se prescriben con el objetivo profiláctico de los antibióticos. El tratamiento antibacteriano intensivo se lleva a cabo en todos los pacientes durante la aparición de una enfermedad infecciosa. Los pacientes no están contraindicados la inmunización de rutina. La terapia de inmunoglobulina de reemplazo está contraindicada cuando el paciente detecta autoantibodes contra IgA. Es necesario tener en cuenta el hecho de que la deficiencia electoral de la IGA se refiere a los defectos inmunes primarios no corrosivos. Las medidas terapéuticas se reducen a la terapia sintomática de enfermedades infecciosas, alérgicas y autoinmunes. Los medicamentos inmunotrópicos se prescriben principalmente debido a la manifestación del aumento de la incidencia infecciosa.
Pronóstico. En pacientes con deficiencia de IgA selectiva, el pronóstico depende de la presencia de un defecto concomitante de anticuerpos específicos, alergias o enfermedades autoinmunes. A menudo, el curso asintomático de la enfermedad puede ser perturbado debido a la acción de los factores externos que afectan, por ejemplo, en una situación estresante, en inmunosupresión, quimioterapia, etc.