Seçici IgA eksikliği en sık görülen birincil immün yetmezlik bozukluğudur (PIDS). Selektif IgA eksikliği olan hastaların insidansı, Kafkasoid popülasyonunda 1:400 ila 1:1000 arasında değişir ve Mongoloid popülasyonunda 1:4000 ila 1:20000 arasında önemli ölçüde daha düşüktür. ABD'de hastalığın prevalansı, çalışma popülasyonunda 223-1000'de 1 ile sağlıklı kan donörlerinde 400-3000'de 1 arasında değişmektedir. Rusya'da bu tür çalışmalar yapılmamıştır.
Bu durum, serum IgA konsantrasyonunun 0,05 g / l'nin altında (dört yaşından büyük çocuklarda) seçici bir azalma ile karakterize edilir. normal seviye diğer serum immünoglobulinleri, normal reaksiyon serum antikorları ve normal hücre aracılı bağışıklık tepkisi. Çoğu çalışmada, erkekler ve kadınlar arasında görülme sıklığı yaklaşık olarak aynıydı.
IgA üretemeyen kişiler, telafi mekanizmaları yoluyla asemptomatik olabilir veya solunum, sindirim veya solunum yollarının sık enfeksiyonlarından muzdarip olabilir. genitoüriner sistem, gastroenterolojik patoloji (örneğin, çölyak hastalığı), saman nezlesi gibi atopik bozukluklara eğilim, bronşiyal astım, atopik dermatit, IgE aracılı gıda alerjisi, nörolojik ve otoimmün hastalıkların yanı sıra (çoğunlukla romatoid artrit, sistemik lupus eritematozus, idiyopatik trombositopenik purpura, Sjögren sendromudur). Seçici IgA eksikliği ile alerjik hastalıklar atopik dermatit ve bronşiyal astım gibi, vakaların %40'ında meydana geldi (Consilium Medicum, 2006). Anafilaktik reaksiyonlar, kan bileşenlerinin transfüzyonu ve bu ürünlerde IgA varlığı ile ilişkili intravenöz immünoglobulinlerin uygulanması sırasında bu hastaların çoğu için tipiktir.
Selektif IgA eksikliğinin klinik semptomları erken çocukluk döneminde ortaya çıkabilir, ancak yaşla birlikte, IgG1 ve G3 alt sınıfları IgM'deki kompansatuar bir artış nedeniyle bulaşan enfeksiyonların sıklığı ve şiddeti azalabilir. Yokluğun başka bir açıklaması klinik semptomlar serum immünoglobulin seviyesindeki azalmaya rağmen normal bir salgı IgA seviyesi olabilir. Ya da tam tersine, başlangıçta seçici IgA eksikliği tanısı konan bazı hastalarda yaygın değişken immün yetmezlik kliniği gelişebilir.
Selektif IgA eksikliği tedavisi şu anda komorbiditelerin belirlenmesinden oluşmaktadır. önleyici tedbirler düşmanlık enfeksiyon riskini azaltmak ve enfeksiyonları hızlı ve etkili bir şekilde tedavi etmek.
Spesifik bir tedavi yoktur. IgA eksikliği olan hastalarda prognoz, ciddi bir hastalık yoksa genellikle iyidir. klinik bulgular. Çocuklarda IgA eksikliği zamanla düzelebilir.
Genetik olarak belirlendiği için, genetik aparattaki kusurlar nedeniyle immün yetmezlik durumları ortaya çıkar. Yaygın değişken immün yetmezliği olan hastalar ve seçici IgA eksikliği olan hastalar sıklıkla aynı ailede bulunur ve ortak bir HLA haplotipini paylaşır; birçoğunda, kromozom 6 üzerinde MCH sınıfı - sınıf 3 içinde nadir aleller ve gen silinmeleri vardır. Son zamanlarda, bazı ailesel yaygın değişken immün yetmezlik ve seçici IgA eksikliği vakalarının, bir TACI (transmembran aktivatörü ve kalsiyum modülatörü ve siklofilin-ligand etkileşimcisi) olarak bilinen protein. TACI mutasyonlarının tespit edilmediği durumlarda, diğer genlerin henüz kaydedilmemiş spontan veya kalıtsal mutasyonlarının, hastalıkların başlamasına neden olması muhtemeldir.
Halihazırda, seçici IgA eksikliğinin olası klinik belirtileri, kurs seçenekleri ve olası eşlik eden hastalıklar yeterince ayrıntılı olarak açıklanmaktadır. Hastalığın tanısında belirleyici, tekrarlanan immünogramlarda normal bir serum immünoglobulin seviyesi olan 4 yaşından küçük çocuklarda IgA'nın serum konsantrasyonunda seçici bir azalmadır. Tedavi, komorbiditelerin belirlenmesinden, enfeksiyon riskini azaltmak için önleyici tedbirler alınmasından ve derhal ve etkili tedavi bulaşıcı hastalıklar.
Rus popülasyonunda bu birincil immün yetmezlik durumunun görülme sıklığı hakkında bilgi yoktur, bu da ülkemizde hastalığın prevalansını benzer çalışmaların halihazırda yürütüldüğü diğer ülkelerle karşılaştırmayı imkansız kılmaktadır.
Ana sorun, seçici IgA eksikliği olan hastaları yönetme taktikleri konusunda birleşik önerilerin olmamasıdır.
Grubun çocukları arasında seçici IgA eksikliğinin görülme sıklığını değerlendirmek için dispanser gözlemi"sık hasta çocuklar" ve klinik belirtilerinin spektrumunu karakterize eder. Rusya Federasyonu Rusya Federasyonu Sağlık Bakanlığı'nın Federal Devlet Bütçe Kurumu "Dmitry Rogachev'in adını taşıyan FNKTs DGOI" ve GBUZ DGKB No. G. N. Speransky DZM bu işi gerçekleştirdi.
Malzemeler ve araştırma yöntemleri
Çalışmanın amacı, GBUZ Çocuk Şehir Klinik Hastanesi No. 2'de gözlenen seçici IgA eksikliği olan çocuklardı. G.N. Speransky DZM. Ayrıca, 2003-2010 dönemi için tıbbi kayıtların geriye dönük bir analizi yapılmıştır. Dispanser gözlem grubundan 9154 hasta "sık hasta çocuklar" (Tablo 1-3).
Muayene sırasında aşağıdaki yöntemler kullanılmıştır:
- klinik ve anamnestik;
- genel ve biyokimyasal analizler kan;
- nefelometri ve akış sitometrisi ile kan bileşiminin immünolojik çalışması;
- skarlaşma testleri;
- immünoblotlama ile spesifik IgE'nin belirlenmesi;
- dış solunum fonksiyonunun incelenmesi;
- rinositolojik çalışma.
Selektif IgA eksikliği tanısı, serum IgA konsantrasyonunun 0,05 g/l'nin altına selektif azalması temelinde konuldu. normal tekrarlanan immünogramlarda diğer serum immünoglobulinleri ve diğerlerinin dışlanması olası nedenler 4 yaşından büyük çocuklarda yetersizlikleri.
Anamnez toplarken Özel dikkat klinik belirtilerin sıklığı ve spektrumu verildi, eşlik eden patoloji ve ayrıntılı bir aile öyküsü. Çocukların klinik muayenesi genel kabul görmüş yöntemlere göre yapıldı. Serumdaki A, G, M, E sınıflarının immünoglobulinlerinin içeriği, bir Dade Behring kiti kullanılarak bir BN 100 nefelometre (Dade Bering, Almanya) üzerinde nefelometri ile belirlendi. Lenfositlerin fenotiplenmesi, floresan olarak etiketlenmiş monoklonal antikorlar Simultest (Becton Dickenson, ABD) kullanılarak bir FacsScan cihazında (Becton Dickenson, ABD) akış sitometrisi ile gerçekleştirildi. Herhangi bir atopi belirtisi olan hastalar ve tüm hastalar artan seviye Nefelometri ile bağışıklık durumu parametrelerinin değerlendirilmesi sonucunda tespit edilen IgE, 4 yaşından büyük çocuklarda skarlaşma testleri yöntemiyle veya çocukların kan serumunda spesifik IgE belirleme yöntemiyle alerjik ek inceleme yapıldı. 4 yaşın altındaki hastalar. Yerleşik bir "bronşiyal astım" tanısı veya bronko-obstrüktif sendrom öyküsü olan çocuklara, Spirovit SP-1 (Schiller AG, İsviçre) aparatı kullanılarak dış solunumun işlevi hakkında bir çalışma yapıldı. Ayrıca mevcut şikayetler de dikkate alınarak ilgili uzmanların gerekli tüm ek muayeneleri ve istişareleri yapılmıştır.
Sonuçlar ve tartışması
"Tekrarlayan akut solunum yolu viral enfeksiyonları", "FIC", "FBR" ve "EBD" sevk tanılı hastaların tıbbi kayıtlarının geriye dönük analizi, bu çocuk grubunda seçici IgA eksikliği sıklığının belirlenmesini mümkün kılmıştır. nüfusa göre iki hatta üç kat daha fazladır.
Mutlak sayı ve yıllara göre bu birincil immün yetmezliğe sahip çocukların yüzdesi Tabloda görülebilir. dört.
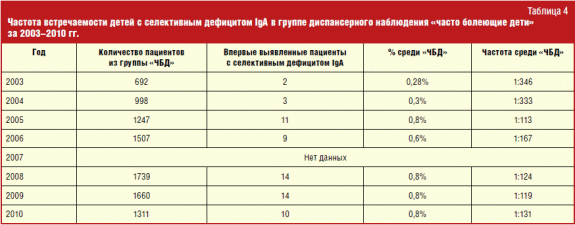
Ne yazık ki, 2007 verileri mevcut değildir. 2003 ve 2004'te 692 ve 998 çocuğa danışıldı. Bunlar arasında, popülasyon ortalamasından biraz daha yaygın olan seçici IgA eksikliği olan toplam 5 hasta tespit edildi - 1:400-600'e karşı sırasıyla 1:346 ve 1:333. 2005'ten bu yana, bu PIDS'li yeni teşhis edilen hastaların sıklığı çarpıcı bir şekilde arttı: 2005'te 1:113, 2006'da 1:167, 2008'de 1:124, 2009'da 1:119 ve son olarak 2010'da 1:131. Çalışmada, görülme sıklığı 2003 yılında 1:346 iken, önceki yıllara göre en yüksek olduğu 2010 yılında 1:131 olarak değişmiştir. İşe başladıktan sonraki üçüncü yılda seçici IgA eksikliği olan hastaların insidansındaki artış, doktorların bu patolojiye ilişkin artan uyanıklığı ve iyileşme ile ilişkilendirilmelidir. laboratuvar teşhisi. Ebeveynleri onları immünologa şikayetleriyle getiren çocukların akışı nedeniyle, doktorların bu hastalık hakkındaki bilgilerini genişletmeye devam etmek gerekir. sık görülen hastalıklar, yıldan yıla artar.
Bu çalışma sırasında 235 çocuk ve 32 yetişkin de ileriye dönük olarak incelendi.
Ana grup, seçici IgA eksikliği teşhisi konan 73 çocuktan oluşuyordu.
İkinci hasta grubu idiyopatik trombositopenik purpuralı (ITP) 153 çocuğu içeriyordu. ITP'li hastaların bağışıklık durumlarının değerlendirilmesi, aralarındaki seçici IgA eksikliğini belirlemek için yapıldı, çünkü bu korelasyon dünya literatüründe tanımlandı ve aynı veriler sırasında aynı veriler elde edildi. bu çalışma. Aralarında IgA bulunmayan tek bir çocuk tespit etmedik. ITP'li hastaların bağışıklık durumunu incelerken, aralarında seçici IgA eksikliği tespit edemememize rağmen, diğer küçük hümoral kusurlar tespit edildi: IgG alt sınıflarının eksikliği, infantil hipogamaglobulinemi, IgA'da kısmi azalma.
Üçüncü grup, seçici IgA eksikliği olan hastaların en yakın akrabaları olan ve aile vakalarını bulmak ve tanımlamak için bağışıklık durumu değerlendirmesi yapılan 20 ila 54 yaş arası 32 yetişkin ve 4 ila 10 yaş arası 8 çocuktan oluşuyordu.
Elde edilen verilerin anketi ve analizi sırasında aşağıdaki sonuçlara ulaşılmıştır.
Selektif IgA eksikliği olan hastalarda erkek ve kadın oranı yaklaşık olarak aynıydı. 40 erkek ve 33 kız çocuğu muayene edildi. Bu, dünya literatürünün verileriyle uyumludur.
Seçici IgA eksikliğinin tespitinin zirvesi 4-7 yaşlarındaydı. Tekrarlayan enfeksiyonlar genellikle Erken yaş veya bir okul öncesi kuruma katılmaya başlamasıyla. Kural olarak, bir immünoloğa gitmeden önce, çocuklar belirli bir bulaşıcı anamnez biriktirdiler, çünkü PIDS'den şüphelenmelerini mümkün kılan belirli işaretler var. Ayrıca, çalışma daha erken yaşta yapılmış olsa ve 4 yıla kadar IgA'nın yokluğunu ortaya çıkarsa bile, bu kesin bir PIDS teşhisi yapmamıza izin vermedi, immünoglobulin sentez sisteminin olgunlaşmamışlığını tamamen dışlayamadık. . Bu nedenle 4 yaşına kadar sorularla tanı konuldu ve dinamiklerde gözlem önerildi. Dolayısıyla sırasıyla 4-7 yıl aralığı.
Selektif IgA eksikliği olan çocuklarda başvuru sırasındaki şikayetlerin başında sık solunum yolu geliyordu. viral enfeksiyonlar karmaşık olmayan bir kursla. Tekrarlayan solunum yolu hastalıklarının başlangıcı, kural olarak, 3 yaşına kadar düştü. Bu da dünya literatür verileriyle örtüşmektedir. Çalışmamızdaki hastaların çoğunluğunun dinamik takibi uzun bir süre, birkaç yıl, bazen hastanın erişkin ağına geçişinden önce yapıldığından, bulaşan enfeksiyonların sıklığının ve şiddetinin azaldığı söylenebilir. yaş. Muhtemelen bu, IgG1 ve IgG3, IgM alt sınıflarının antikorlarındaki telafi edici bir artıştan kaynaklanıyordu, ancak bu konu daha fazla çalışma gerektiriyor. Tedavi sırasında en sık görülen ikinci şikayet, komplikasyonlarla ortaya çıkan sık akut solunum yolu viral enfeksiyonlarıydı. Dinamik gözlem ile gösterildiği gibi, hastalarımızda yaşla birlikte komplike, atipik olarak ortaya çıkan akut solunum yolu viral enfeksiyonlarının sıklığı da azalmıştır.
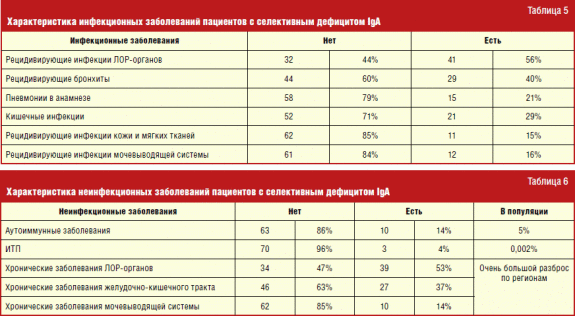
Seçici IgA eksikliği olan hastalarda bulaşıcı hastalıklar yelpazesinden, önde gelen yer KBB organlarının bulaşıcı hastalıkları ve alt organların enfeksiyonları tarafından işgal edildi. solunum sistemi. Bunun nedeni, yerel bağışıklığın bir parçası olan salgı IgA'daki bir azalmanın, temasa en duyarlı olan mukoza zarlarında mikroorganizmaların kolay enfeksiyonuna ve çoğalmasına yol açmasıdır. bulaşıcı hastalıklar havadaki damlacıklar tarafından iletilir.
Bulaşıcı olmayan hastalıklar yelpazesinde, selektif IgA eksikliğinin en önemli belirtileri olan otoimmün hastalıklarla, özellikle idiyopatik trombositopenik purpura (100 binde 1.5-2) ile bariz bir korelasyon bulundu.
İtibaren otoimmün hastalıklar seçici IgA eksikliği olan hastalarda, juvenil romatoid artrit (4 kez), kronik idiyopatik trombositopenik purpura (3 kez), otoimmün hepatit (3 kez) en yaygındı. Ayrıca dünya literatürüne göre selektif IgA eksikliği olan hastalarda akrabalar arasında otoimmün hastalıkların görülme sıklığında artış vardır. Ancak bizim çalışmamıza göre sayıları genel popülasyon değerlerini geçmemiştir.
Selektif IgA eksikliği olan hastalarda atopik hastalıkların sıklığı genel popülasyona göre anlamlı derecede yüksekti (Tablo 4). Sadece alerjik rinit sıklığı genel popülasyonla karşılaştırılabilir. Benzer gözlemler daha önce yapılan bir dizi çalışmada yansıtılmıştır. IgA eksikliği olan hastaların çoğunda alerjik hastalıkların, bu immünolojik bozukluğu olmayan kişilere göre daha şiddetli olduğu söylenemez. Bununla birlikte, yüksek atopi prevalansı, henüz klinik olarak ortaya çıkmamış selektif IgA eksikliği formlarını tanımlamak için immünolojik bir inceleme yapılması sorununu ortaya çıkarmaktadır. Bu, mevcut atopik durum için tedaviye yaklaşım açısından belirleyici bir role sahip olmasa da, seçici IgA eksikliği olan kişiler için zamanında tanı koymaya ve olası riskleri azaltmaya yardımcı olacaktır.
Laboratuvar parametrelerindeki kalıcı değişiklikler nedeniyle seçici IgA eksikliği olan çocuklarda dinamik gözlem sırasında tekrarlanan immünogramları analiz ederken, iki büyük gruplar hastalar. A grubunda başka bir değişiklik olmaksızın IgA'nın yokluğu not edildi. B grubunda, IgA'nın yokluğu, IgG seviyelerinde kalıcı bir artış ile birleştirildi. Bu hasta gruplarının karşılaştırmalı bir analizi yapıldı.

Bu gruplarda klinik belirtilerin başlama yaşı önemli ölçüde farklılık göstermedi.
Seçici IgA eksikliği olan hastalarda, IgG seviyelerindeki bir artışın, deri ve yumuşak dokuların tekrarlayan enfeksiyöz hastalıkları ile korele olduğu bulundu. Bu konu daha fazla çalışma gerektirir.
Bu hasta grupları karşılaştırıldığında, alerjik patoloji spektrumunda anlamlı bir farklılık yoktu.
Çalışma sırasında, seçici IgA eksikliği olan 20 hasta ailesinde bağışıklık durumu değerlendirildi. 4 ailevi vaka tespit edildi. Ayrıca ayrıntılı bir aile öyküsü alındı. Ağırlaştırılmış bulaşıcı anamnezi olan ve muayene yapmayı başaran yetişkin akrabalar arasında, bazı hümoral bağışıklık ihlalleri vardı. Buna göre, küçük hümoral defektler (özellikle seçici IgA eksikliği) tespit edildiğinde, özellikle ağırlaştırılmış enfeksiyon öyküsü varlığında yakın akrabanın muayenesi zorunludur.
Dispanser gözlem grubundaki çocuklar arasında seçici IgA eksikliğinin "sık hasta çocuklar" genel çocuk popülasyonundan çok daha sık meydana gelmesi nedeniyle, pratisyen pediatristlerin bu hastalık hakkında uyanık olmaları gerekir. Klinik belirtiler çok değişken olduğundan şüphelenmek her zaman kolay değildir: asemptomatik formlardan sık sık ihtiyaç duyulan tekrarlayan bakteriyel enfeksiyonlara kadar. antibiyotik tedavisi. Çocuk doktorlarının ve ayaktan ve yatan hasta bağlantısının dar uzmanlarının, bağışıklığın hümoral bağlantısındaki küçük kusurlar hakkındaki bilgilerinin genişletilmesi önerilir.
Selektif IgA eksikliği olan hastalarda alerjik patoloji (bronşiyal astım, atopik dermatit, besin alerjisi) görülme sıklığı anlamlı olarak daha yüksek olduğundan, otoimmün hastalıkların ve hematolojik hastalıkların görülme sıklığının yanı sıra görülme sıklığı da daha fazladır. kronik hastalıklar(KBB organları, genitoüriner sistem, gastrointestinal sistem) popülasyonda olduğundan daha eksiksiz ve zamanında sağlanması için kimliğinin belirlenmesi zorunludur. Tıbbi bakım hastalar.
Yüklü enfeksiyon öyküsü olan çocukların, hematolojik ve otoimmün hastalıkları olan hastaların bir immünolog/immünolojik muayene ile konsültasyona yönlendirilmesi ve alerjik hastalığı olan hastalarda total IgA seviyesinin tarama incelemesi yapılması önerilir.
Çalışma, seçici IgA eksikliği olan çocukların çoğunluğunun, IgA eksikliğinin varlığının bir korelasyon gösterdiğini buldu. otoimmün patoloji ve tekrarlanan immünogramlarda IgG'de kalıcı bir artış. Diğer hastalıklar için böyle bir ilişki bulunamadı. Göstergelerdeki bu tür değişiklikler, bir çocukta otoimmün patolojinin gelişimi için bir risk faktörüdür ve özel dikkat gerektirir.
Ailede selektif IgA eksikliği öyküsü varlığı ile hastalardaki klinik bulguların şiddeti arasında bir ilişki kurulmamış olmakla birlikte, bu hastalarda özellikle ağırlaştırılmış enfeksiyon öyküsü varlığında yakın akrabanın muayenesi zorunludur. .
Edebiyat
- Hammarstrom L., Lonnqvist B., Ringden O., Smith C.I., Wiebe T. Aplastik anemili kemik iliği aşılı bir hastaya IgA eksikliğinin transferi // Lancet. 1985; 1 (8432): 778-781.
- Latiff A.H., Kerr M.A.İmmünoglobulin A eksikliğinin klinik önemi // Klinik Biyokimya Annals. 2007; 44 (Pt 2): 131-139.
- Al-Attas R.A., Rahi A.H. Araplarda birincil antikor eksikliği: Doğu Suudi Arabistan'dan ilk rapor // Klinik İmmünoloji Dergisi. 1998; 18(5): 368-371.
- Carneiro-Sampaio M.M., Carbonare S.B., Rozentraub R.B., de Araujo M.N., Riberiro M.A., Porto M.H. Brezilyalı kan bağışçıları ve sağlıklı hamile kadınlar arasında seçici IgA eksikliğinin sıklığı // Allergology Immunopatology (Madr). 1989; 17(4):213-216.
- Ezeoke A.C. Doğu Nijerya'da Seçici IgA eksikliği (SIgAD) // Afrika Tıp ve Tıp Bilimleri Dergisi. 1988; 17(1):17-21.
- Feng L.Çin'deki 6 millet arasında seçici IgA eksikliğinin epidemiyolojik çalışması // Zhonghua Yi Xue Za Zhi. 1992; 72(2): 88-90, 128.
- Pereira L.F., Sapina A.M., Arroyo J., Vinuelas J., Bardaji R.M., Prieto L.İspanya'da seçici IgA eksikliğinin yaygınlığı: düşündüğümüzden daha fazla // Kan. 1997; 90(2): 893.
- Wiebe V., Helal A., Lefranc M.P., Lefranc G. T17 immünoglobulin CH multigen silmesinin moleküler analizi (del A1-GP-G2-G4-E) // İnsan Genetiği. 1994; 93(5):5.
L.A. Fedorova*,
E. S. Pushkova*
I.A. Korsunsky** , 1 ,Tıp Bilimleri Adayı
A.P. Prodeus*,tıp bilimleri doktoru, profesör
İmmün yetmezlik - ana bileşenlerin nicel göstergelerinde ve / veya fonksiyonel aktivitesinde azalma bağışıklık sistemi, vücudun patojenik mikroorganizmalara karşı savunmasının ihlaline yol açar ve artan bulaşıcı morbidite ile kendini gösterir.
Bildiğiniz gibi, bağışıklık sisteminin ana işlevi, vücuda giren antijenik nitelikteki yabancı maddelerin tanınması ve ortadan kaldırılmasıdır. çevre(mikroorganizmalar) veya endojen (tümör hücreleri). Bu işlev, faktörler kullanılarak uygulanır. doğuştan gelen bağışıklık(fagositoz, antimikrobiyal peptitler, tamamlayıcı sistem proteinleri, NK hücre sistemleri, vb.) ve hücresel ve hümoral bağışıklık tepkileri kullanılarak gerçekleştirilen edinilmiş veya edinilmiş bağışıklık. Vücudun bağışıklık savunmasının bileşenlerinin aktivitesinin düzenlenmesi ve bunların etkileşimi, sitokinler ve hücreler arası temasların yardımıyla gerçekleşir.
Bağışıklık sisteminin listelenen bileşenlerinin her birinde ve bunların düzenlenmesi mekanizmalarında, ana klinik tezahürü bulaşıcı hastalıkların patojenlerine karşı artan duyarlılık olan bağışıklık yetmezliğinin gelişmesine yol açan rahatsızlıklar meydana gelebilir. 2 tip immün yetmezlik vardır: birincil ve ikincil.
Birincil immün yetmezlikler(PID) - kalıtsal hastalıklar bağışıklık tepkisini kontrol eden genlerdeki kusurlardan kaynaklanır. PID, bağışıklık kusurlarının, klinik belirtilerin ve moleküler bozuklukların doğası ve şiddeti bakımından çeşitlilik gösteren bir hastalıktır. PID'nin klinik tablosu, çoğunlukla bronkopulmoner sistemin tekrarlayan ve kronik, şiddetli enfeksiyöz süreçleri ile karakterizedir.
ve KBB organları, deri ve mukoza zarları; pürülan lenfadenit, apse, osteomiyelit, menenjit ve sepsis gelişebilir. Bazı formlarda alerji, otoimmün hastalıklar ve bazılarının gelişimi vardır. malign tümörler. gecikmesine dikkat edilmelidir. yaş göstergeleri fiziksel Geliştirme. Şu anda yaklaşık 80 PID tanımlanmıştır ve bu hastalıkların çoğunun gelişiminden sorumlu genler tanımlanmıştır. Yeterli laboratuvar analizleri, patolojiyi lenfositler düzeyinde ve patolojiyi lenfosit dışı yıkım mekanizmaları ve antijenlerin atılımı düzeyinde ayırt etmeyi mümkün kılar.
PID'nin yaygınlığı hastalığın formuna bağlıdır ve ortalama 1:10.000 ila 1:100.000 yenidoğan arasındadır. seçici kıtlıkÖrneğin IgA, genel popülasyonda 1:500 ila 1:1500 kişi arasında çok daha yaygındır. yaygınlık çeşitli formlar PID değişir Farklı ülkeler. Antikor üretiminde en yaygın kusurlar - vakaların %50-60'ı, kombine PID - %10-30, fagositoz kusurları - %10-20, kompleman kusurları - %1-6. Yaygın değişken immün yetmezlik (CVID) gibi bazı PID biçimleri daha sonra başlasa da, çoğu PID erken çocukluk döneminde ortaya çıkar.
Gelişim mekanizmalarına göre, 4 ana PID grubu ayırt edilir:
Grup 1 - ağırlıklı olarak hümoral veya B-hücresi
PID;
grup 2 - kombine PID (tüm T hücresi immün yetmezliklerinde B hücrelerinin işlevinin ihlali vardır);
grup 3 - fagositozdaki kusurların neden olduğu PID;
4. grup - kompleman sistemindeki kusurların neden olduğu PID.
Primer immün yetmezliklerin tanı prensipleri
Erken tanı ve tedaviye zamanında başlanması hastalığın prognozunu belirler. Bölge çocuk doktorları düzeyinde tanı koymak, genellikle hastanın bir immünolog tarafından zamanında konsültasyonunun olmaması ve özel bir laboratuvar immünolojik muayenesinin yapılmasından kaynaklanan bazı zorluklar sunar (Tablo 11-1). PID'nin klinik tablosunun özellikleri ve değişiklikleri hakkında bilgi sahibi olmasına rağmen
Genel klinik laboratuvar testlerindeki bulgular, PID'den şüphelenmeyi ve hastayı uzmanlara sevk etmeyi mümkün kılar. Avrupa İmmün Yetmezlikler Derneği, PID'nin erken teşhisi için protokoller geliştirmiş ve ayrıca elektronik veritabanı Avrupa PID kaydı verileri. PID diyagnostik algoritması, Şek. 11-1.
Tablo 11-1.Şüpheli immün yetmezlik için immünolojik muayenenin aşamaları
Pirinç. 11-1. Primer immün yetmezliklerin teşhisi için algoritma
Primer immün yetmezliklerin klinik tablosunun genel özellikleri
lider klinik tablo PID sözde bulaşıcı sendrom- genel olarak bulaşıcı hastalıkların patojenlerine karşı artan duyarlılık, alışılmadık derecede şiddetli tekrarlayan (tekrarlayan) klinik kursu, atipik patojenlerin hastalığının etiyolojisindeki varlığı (genellikle fırsatçı). Patojenin tipi, bağışıklık kusurunun doğasına göre belirlenir. Antikor oluşumundaki kusurlarla, antikorlara karşı direnci belirlemek mümkündür. antibakteriyel ilaçlar flora - stafilokok, streptokok, Haemophilus influenzae. T hücresi bağışıklık eksikliğinde, bakterilere ek olarak virüsler (örneğin, herpesvirüs ailesi), mantarlar tespit edilir. (Candida spp., Aspergillus vb.) ve fagositik kusurlarla - stafilokoklar, gram-negatif bakteriler, mantarlar, vb.
Laboratuvar araştırması
Klinik bulgular PID'yi düşündürürse, aşağıdaki incelemeler yapılmalıdır:
Ayrıntılı bir kan formülünün belirlenmesi (lenfositlerin nicel ve yüzde göstergeleri özellikle önemlidir);
Kan serumunda IgG, IgA ve IgM düzeylerinin belirlenmesi;
T- ve B-lenfositlerin alt popülasyonlarının sayılması;
İle özel göstergeler:
◊ fagositlerin fonksiyonel durumunun analizi (en basit ve en bilgilendirici analiz, tetrazolium mavisinin restorasyonu için yapılan testtir);
◊ tamamlayıcının ana bileşenlerinin içeriği için analiz (C3 ve C4 ile başlayın);
◊ HIV enfeksiyonu testi (olası risk faktörleri varsa);
◊ endike olduğunda moleküler genetik çalışmalar.
Primer immün yetmezliklerin tedavi prensipleri
PID tedavisinin temel amacı, hastalığın komplikasyonlarının tedavisi ve önlenmesidir. Bu yaklaşım, PID'deki bağışıklık sistemindeki kusurların genetik düzeyde ortaya konmasından kaynaklanmaktadır. Şu anda, üzerinde yoğun araştırmalar yürütülmektedir.
tedavilerinde daha radikal yöntemlerin ortaya çıkmasına neden olabilecek immün yetmezliklerin tedavisi.
PID formuna bağlı olarak tedavi, ikame tedavisi, hastalığın enfeksiyöz, otoimmün belirtilerinin tedavisi ve önlenmesi, malign neoplazmların tedavisi ve kullanımından oluşur. özel yöntemler hematopoietik kök hücre nakli dahil (PID tipine bağlı olarak).
İMMÜNOGLOBÜLİNLERDEKİ KUSURLAR
Çocuklarda geçici hipogamaglobulinemi
Çocuklarda geçici hipogamaglobulinemi, immünoglobulin sisteminin kademeli oluşumunun fizyolojik bir özelliği ile ilişkilidir. IgM ve IgA antikor oluşumunun olgunlaşması büyük ölçüde "geciktirilir". Sağlıklı çocuklarda maternal IgG içeriği giderek azalır ve altı ay sonra kendi IgG antikorlarının üretimi artar. Ancak bazı çocuklarda immünoglobulin seviyelerindeki artış gecikir. Bu tür çocuklar tekrarlayan bakteriyel enfeksiyonlardan muzdarip olabilir. Bu durumlarda, donör immünoglobulin preparatlarının infüzyonlarına (intravenöz immünoglobulin verilmesi) başvurulmamalıdır.
Seçici immünoglobulin A eksikliği
İmmünoglobulin A'nın seçici eksikliği (SD IgA - IgA'nın Seçici Eksikliği) bir gen kusurunun sonucu olarak gelişir tnfrsf13b
veya r). Diğer sınıfların immünoglobulinlerinin varlığında IgA eksikliği, genel popülasyonda 1:500-1500 kişi sıklığında (alerjisi olan hastalarda, hatta daha sık) tespit edilen en yaygın immün yetmezliktir. Seçici IgA eksikliği vardır, yani. alt sınıflardan birinin eksikliğinden (vakaların %30'u) ve tam (vakaların %70'i) oluşur. IgA2 alt sınıfının eksikliği, IgA1 alt sınıfının eksikliğinden daha belirgin bir klinik tabloya yol açar. IgA eksikliğinin diğer bozukluklarla kombinasyonları da mümkündür: IgG biyosentezinde bir kusur ve T-lenfosit anormallikleri ile. Seçici olan bireylerin büyük çoğunluğu
IgA eksikliği pratik olarak sağlıklıdır. 2 yaşın altındaki çocuklar için IgA eksikliği fizyolojik bir durumdur.
Serum IgA konsantrasyonunda bir düşüş tespit edin.<5 мг/дл у детей старше 4 лет; IgG и IgM в норме, количество и соотношение субпопуляций лимфоцитов и их функциональная активность могут быть в норме.
klinik tablo. IgA eksikliği ile 3 grup patolojik sendrom gelişebilir: bulaşıcı, otoimmün ve alerjik. IgA eksikliği olan hastalar, üst solunum yolu ve sindirim organlarının tekrarlayan bulaşıcı hastalıklarına yatkındır. En sık ve şiddetli olan çeşitli otoimmün hastalıklardır (romatoid artrit, ankilozan spondilit, Sjögren sendromu, beyin damarlarına zarar veren vaskülit, otoimmün tiroidit, SLE, glomerülonefrit, hemolitik anemi, tip I diyabetes mellitus, vitiligo, vb.). Çölyak hastalığı insidansı, normal IgA'lı çocuklarda 10 kat daha fazladır. En sık saptanan alerjik belirtiler inek sütü protein intoleransı, atopik dermatit (AtD), bronşiyal astımdır.
Tedavi. Asemptomatik vakalar herhangi bir özel tedavi gerektirmez; bulaşıcı, otoimmün ve alerjik hastalıkların klinik belirtilerinin varlığında, standartlara uygun olarak tedavi yapılır.
Alıcıda IgA'ya karşı antiizotipik antikorların oluşma ve bunların neden olduğu transfüzyon komplikasyonlarının gelişme olasılığı yüksek olduğundan, ne seçici ne de tam IgA eksikliği için donör immünoglobulinleri ile ikame tedavisi endike değildir.
Agammaglobulinemi B hücre eksikliği ile
X'e bağlı agamaglobulinemi (Bruton hastalığı) tüm agamaglobulinemi vakalarının %90'ını oluşturur. Kusurlu genin taşıyıcılarının erkek ve oğulları (אּ, ρ) hastalanır btk (Xq21.3-q22), B-lenfosit spesifik protein tirozin kinaz Btk'yi kodlayan (Bruton'un tirozin kinazı- Bruton'un tirozin kinazı). Kusurun bir sonucu olarak, hücre içi sinyal yollarının ihlali, ağır immünoglobulin zincirlerinin rekombinasyonu, farklılaşma vardır.
B-lenfositlerine pre-B hücrelerinin replikasyonu. B hücre eksikliği olan hastaların %10'unda agamaglobulinemi otozomal çekinik olarak kalıtılır. Pre-B hücre reseptörü, sitoplazmik B hücre adaptör proteini (BLNK) ve gen dahil olmak üzere şu ana kadar altı genetik kusur tanımlanmıştır. Lösin-Zengin Tekrar İçeren 8 (LRRC8).
Laboratuvar çalışmalarından elde edilen veriler. Periferik B-lenfositleri yoktur. Kemik iliği sitoplazmada μ-zincirli pre-B hücreleri içerir. T-lenfositlerin sayısı ve T-lenfositler için fonksiyonel testler normal olabilir. Kandaki IgM ve IgA tespit edilemez; IgG mevcut olabilir, ancak küçük miktarlarda (0.4-1.0 g/l). Kan grubu antijenlerine ve aşı antijenlerine (tetanoz, difteri toksinleri vb.) karşı antikor yoktur. Nötropeni gelişebilir. Lenfoid dokunun histolojik incelemesi: Lenfoid foliküllerde germinal (germ) merkezler ve plazma hücreleri yoktur.
klinik tablo. Aile öyküsü bilinmiyorsa ortalama 3,5 yaşında tanı belirgin hale gelir. Hastalık, lenfoid doku hipoplazisi, şiddetli pürülan enfeksiyonlar, üst (sinüzit, orta kulak iltihabı) ve alt (bronşit, zatürree) solunum yollarının bulaşıcı hastalıkları ile karakterizedir; olası gastroenterit, piyoderma, septik artrit (bakteriyel veya klamidyal), septisemi, menenjit, ensefalit, osteomiyelit. Solunum yolu hastalıklarının en yaygın etken maddeleri şunlardır: Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus, ishal bağırsak bakterileri veya giardia Giardia lamblia. Ayrıca agamaglobulinemili hastalar, kronik pnömoni, pürülan artrit, sistit ve deri altı doku apselerinin nedeni olan mikoplazmalar ve üreaplazmaların neden olduğu bulaşıcı hastalıklara karşı hassastır. Virüslerden, nörotropik virüsler ECHO-19 ve coxsackie tipiktir ve hem şiddetli akut hem de kronik ensefalite ve ensefalomiyelite neden olur. Enterovirüs enfeksiyonlarının belirtileri dermatomiyozit benzeri sendrom, ataksi, baş ağrıları ve davranış bozuklukları olabilir. Hasta çocuklarda, canlı çocuk felci aşısı ile aşılama sırasında, kural olarak, poliomyelit virüsünün mukoza zarlarından uzun süreli atılımı, ayrıca, restore edilmiş ve artan virülans ile (yani, çocuk koleksiyonunda -
sağlıklı çocuklarda aşılanmış bağışıklık yetersizliği olan bir çocukla temas sonucunda gerçek bir çocuk felci enfeksiyonu riski yoktur). Agammaglobulinemideki otoimmün bozukluklar, romatoid artrit, skleroderma benzeri sendrom, skleredema, ülseratif kolit, tip I diabetes mellitus (Th1 bağışıklık tepkisinin baskınlığından dolayı) ile temsil edilebilir.
Fiziksel inceleme. Fiziksel gelişimdeki gecikmeye, parmakların şekline (bavul şeklinde parmaklar), göğüs şeklindeki değişikliklere, alt solunum yolu hastalıklarının karakteristiğine, lenf düğümlerinin ve bademciklerin hipoplazisine dikkat edin.
Tedavi.
Yerine koyma tedavisi: Yaşam boyunca her 3-4 haftada bir intravenöz immünoglobulin preparatları uygulanır. İmmünoglobulin dozları, hastanın serumunda yaş normunun alt sınırıyla örtüşen konsantrasyonlarını oluşturacak şekilde seçilir.
Gen Tedavisinin Olanağının Tartışılması - Gen btk klonlanmıştır, ancak aşırı ekspresyonu hematopoietik dokunun malign transformasyonu ile ilişkilidir.
Kalıcı nötropeni durumunda büyüme faktörleri kullanılır. Otoimmün patoloji belirtileri ortaya çıktığında, monoklonal antikorların (infliximab, vb.) Yazılması mümkündür.
Ortak Değişken İmmün Yetmezlik
Yaygın değişken immün yetmezlik (CVID), antikor sentezi ve hücresel immünitede bir kusur ile karakterize edilen bir grup sendromdur. CVID için güvenilir bir tanı kriteri, aşağıdaki belirtilerden biriyle kombinasyon halinde her iki cinsiyette iki veya üç ana izotipin immünoglobulin içeriğinde önemli bir azalmadır:
2 yaş üzeri hastalığın başlangıcı;
İzohemaglütininlerin yokluğu ve/veya aşıya düşük yanıt;
Agammaglobulineminin diğer nedenlerinin dışlanması.
Bazı hastalarda CVID gelişiminin nedeni, B hücrelerinin olgunlaşma ve hayatta kalma süreçlerinde yer alan molekülleri kodlayan genlerdeki mutasyonlardır: BAFF-R (B-hücresi Aktivasyon Faktörü Reseptörü), keşif balonu-1 (B-lenfosit kaynaklı olgunlaşma proteini-1) ve ICOS (İndüklenebilir kostimülatör). B-lenfositlerin plazma hücrelerine farklılaşma yeteneğinin ihlali vardır, antikor oluşumunda kusurlar gelişir, T-lenfositlerin işlevsizliği mümkündür ve bulaşıcı hastalıklara karşı artan bir duyarlılık gözlenir. Sendrom erken çocukluk, ergenlik veya genç erişkinlerde ortaya çıkabilir.
Laboratuvar çalışmalarından elde edilen veriler. IgG ve IgA'nın (hastaların yaklaşık %50'sinde) ve IgM'nin (saptanamayan miktarlara kadar) önemli ölçüde azalması. Kandaki B-lenfosit sayısı normaldir veya azalmıştır. Çoğu hastada T-lenfosit sayısı normaldir. Şiddetli hastalarda lenfopeni gelişebilir (1 litre kan başına 1500x103 hücreden az). NK hücrelerinin sayısı azalır. Bağışıklamaya yanıt olarak spesifik antikorların üretimi azalır veya yoktur. Mitojenlerin ve antijenlerin etkisi altında lenfositlerin proliferasyonu ve IL-2 oluşumu önemli ölçüde bozulur.
klinik tablo. Tekrarlayan bakteriyel enfeksiyon hastalıkları, esas olarak solunum yolu ve paranazal sinüslerde lokalizasyon ile tespit edilir. Tanı anında solunum yolu enfeksiyonları bronşektazi ve yaygın akciğer dokusu lezyonlarına kadar ilerleyebilir. Belki de ishal, steatore ve malabsorpsiyon (ve buna bağlı olarak kilo kaybı) ile kendini gösteren sindirim sisteminin bulaşıcı bir lezyonu. Genellikle enfeksiyonlar neden olur Giardia lamblia, Pneumocystis carinii veya ailenin virüsleri Herpetoviridae. CVID'li hastalar, mikoplazmalar ve üreaplazmaların neden olduğu pürülan artrit gelişimine eğilimlidir. Ensefalomiyelit, çocuk felci ve dermatomiyozit benzeri sendromlar, deri ve müköz membran lezyonları enterovirüs enfeksiyonlarının belirtileri olabilir. otoimmün hastalıklar şiddetlidir ve CVID'nin prognozunu belirleyebilir. Bazen CVID'nin ilk klinik belirtileri artrit, ülseratif kolit ve Crohn hastalığı, sklerozan kolanjit, malabsorpsiyon, SLE, nefrit, miyozit, lenfoid interstisyel pnömoni, nötropeni şeklinde otoimmün akciğer hasarı,
trombositopenik purpura, hemolitik anemi, pernisiyöz anemi, total alopesi, retinal vaskülit, ışığa duyarlılık. CVID'li hastalarda sarkoidoz benzeri granülomların sıklığı (vakaların %15'inde) ve malign olmayan lenfoproliferasyon önemli ölçüde artar. Tedavi.
Antibakteriyel kemoterapi.
Yerine koyma tedavisi: Yaşam boyunca her 3-4 haftada bir intravenöz immünoglobulin preparatları uygulanır.
Otoimmün komplikasyonlarla - immünosupresif tedavi (glukokortikoidler, azatioprin, siklosporin A) ve monoklonal antikorların (infliximab, vb.) atanması mümkündür.
Hiper-IgM sendromları
Hiper-IgM sendromları, IgG, IgA ve normal veya yüksek serum IgM konsantrasyonlarında belirgin bir azalma veya tam yokluk ile karakterize oldukça nadir görülen hastalıklardır. Bunun nedeni, B-lenfositlerinin immünoglobulin sınıf geçişi ve değişken alan hipermutajenezi gerçekleştirememesidir. Bugüne kadar, hiper-IgM sendromunun gelişmesine yol açan 6 genetik kusur tanımlanmıştır.
. Tip 1 (HIGM 1). CD40 ligandının X'e bağlı eksikliği (hiper-IgM sendromlarının vakalarının %70'i), T hücrelerinin B lenfositleri ile etkili bir şekilde etkileşime girememesiyle sonuçlanır.
. Tip 2 (HIGM 2). Otozomal resesif, sitidin deaminazın (gen Aicda, 12p13)- immünoglobulinler ve hipermutajenez sınıfları arasında geçiş yapan bir enzim.
. Tip 3 (HIGM 3). Otozomal resesif, CD40 molekül genindeki bir mutasyonla ilişkili. Aynı zamanda, B-hücrelerinin kendileri, T-lenfositleri ile etkili bir şekilde etkileşime giremezler. Fenotipik belirtiler tip 1'dekine benzer.
. Tip 4 (HIGM 4). Otozomal çekinik; bazı durumlarda mutasyonlar meydana gelir de novo. UNG - urasil-DNA glikosilazdaki bir kusurla ilişkili - aynı zamanda dahil olan bir enzim
immünoglobulin sınıflarını değiştirirken, ancak AID'nin etkisinden sonra. Bu durumda hipermutajenez etkilenmez ve sendrom daha az şiddetlidir.
. Tip 5 (HIGM 5). Kusur sadece sınıf geçişindedir, hipermutajenez etkilenmez. Nedensel mutasyon henüz tanımlanmadı, ancak sonrasında etki eden enzimde açıkça bir kusur var.
YARDIM.
. Tip 6 (HIGM-ED). Dishidrotik ektodermal displazi ile ilişkili X'e bağlı, CD40'tan bozulmuş sinyalleşmeye yol açan NEMO (NF-kB modülatörü) eksikliğinden kaynaklanır.
X'e bağlı hiper-IgM sendromu diğerlerinden daha sık tespit edilir. CD40L'yi kodlayan gende (CD154, genin bulunduğu gende) bir kusur ile gelişir. Xq26-q27.2)- CD40 için ligand. T-lenfositler tarafından CD40L ekspresyonunun yetersizliği, B-lenfositlerdeki immünoglobulin sınıflarının IgM'den diğer izotiplere geçişinin imkansızlığının yanı sıra, hafıza B hücrelerinin, T-hücre repertuarının ve yönlendirilmiş Thl-hücre yanıtının bozulmuş oluşumuna yol açar. hücre içi mikroorganizmalara karşı çocuklar hasta olsun
Laboratuvar çalışmalarından elde edilen veriler. IgG, IgA, IgE belirlenemez veya çok küçük miktarlarda tespit edilir. IgM seviyesi normaldir (vakaların %50'sinde) veya genellikle önemli ölçüde yükselmiştir. T ve B hücrelerinin sayısı normaldir; antijenler tarafından indüklenen T hücrelerinin azaltılmış proliferatif tepkisi. IgM poliklonal, bazen monoklonaldir. IgM izotipinin otoantikorları tespit edilir (antieritrosit, antiplatelet, antitiroid, düz kas dokusu antijenlerine karşı antikorlar). Lenfoid dokuda germinal merkez yoktur, ancak plazma hücreleri vardır.
klinik tablo.İlk belirtiler bebeklik ve erken çocukluk döneminde ortaya çıkar. Tekrarlanan ile karakterize enfeksiyonlar fırsatçı (nedeniyle oluşan) dahil olmak üzere farklı lokalizasyon (öncelikle solunum yolu) Pneumocystis carini). Virüs enfeksiyonları (sitomegalovirüs ve adenovirüsler) de karakteristiktir, Criptococcus neoformans, mikoplazmalar ve mikobakteriler. Cryptosporidial enfeksiyon, akut ve kronik ishale (hastaların %50'sinde gelişir) ve sklerozan kolanjite neden olabilir. Genellikle anemi, nötropeni, oral mukoza ülserasyonu, diş eti iltihabı, ülseratif gelişir.
yemek borusu lezyonları, bağırsağın çeşitli kısımları, ülseratif kolit. yatkınlık gösterir otoimmün bozukluklar(seronegatif artrit, glomerülonefrit, vb.) ve malign neoplazmalar (esas olarak lenfoid doku, karaciğer ve safra yolları). Lenfadenopati, hepato ve splenomegali gelişebilir. Tedavi
İntravenöz immünoglobulin ile düzenli replasman tedavisi.
Antibakteriyel kemoterapi. Pneumocystis pnömonisinin önlenmesi ve tedavisi için ko-trimoksazol [sülfametoksazol + trimetoprim] ve pentamidin kullanılır.
Karaciğer ve safra yollarına zarar vermemek için sadece kaynamış veya filtrelenmiş su kullanmalı, düzenli muayeneler (ultrason, endikasyon varsa karaciğer biyopsisi) yapmalısınız.
Nötropeni ve ağız boşluğunun ülserasyonunun tedavisinde glukokortikoidler ve granülosit koloni uyarıcı faktör müstahzarları kullanılır.
Otoimmün komplikasyonların gelişmesiyle birlikte, immünosupresif tedavi (glukokortikoidler, azatioprin, siklosporin A) ve monoklonal antikorlara dayalı ilaçlar reçete edilir.
Optimal tedavi, HLA uyumlu donörlerden kemik iliği naklidir (hayatta kalma oranı %68, en iyi 8 yaşından önce yapılır).
BİRİNCİL T-LENFOSİT BOZUKLUĞU İLE KOMBİNE İMMÜN YETMEZLİK
Şiddetli kombine immün yetmezlik
TKIN (SCID - Şiddetli Kombine Bağışıklık Yetmezliği)- T-lenfosit seviyesinde bir azalma veya bunların tamamen yokluğu ve adaptif bağışıklığın ihlali ile karakterize edilen bir grup sendrom. . Erken evrelerde lenfoid ve miyeloid progenitörlerin olgunlaşmasının bozulması ile karakterize retiküler disgenezi: nötropeni ve T - B - NK - .
. Gen mutasyonu nedeniyle X'e bağlı SCID IL-2RG[(CD132, toplam de- IL-2, IL-4, IL-7, IL-9, IL-15 ve IL-21 için reseptör zinciri), Xq13.1-q21.1,אּ ], bu, reseptörlerin bloke edilmesine ve hedef hücrelerin karşılık gelen interlökinlerin etkisine yanıt verememesine yol açar (tüm SCID vakalarının %50'sinden fazlası); T - B + NK - .
. Janus3 tirozin kinaz eksikliği [gen JAK3 (19p13.1),ρ ]; gen kusurları ile, aktivasyon sinyalinin genelden iletilmesi de- T ve NK hücrelerinin farklılaşmasının bozulmasına yol açan hücre çekirdeğine IL-2, IL-4, IL-7, IL-9, IL-15, IL-21 zincirleri; T - B+NK - .
. Protein tirozin fosfataz eksikliği (CD45, gen PTPRC, 1q31-q32); gen kusurlu olduğunda, Csk kinazın protein tirozin kinaz Src üzerindeki inhibitör aktivitesi, TCR ve BCR'nin ITAM alanlarının bozulmuş fosforilasyonu ile arttırılır; T - B+NK+.
. İmmünoglobulinlerin V(D)J-segmentlerinin ve TCR [genlerinin rekombinasyonunu aktive eden RAG1 ve RAG2 enzimlerinin tam eksikliği RAG1 ve RAG2 (11r13),ρ ]; T-B-NK+.
. Omenn sendromu (RAG1 enzimlerinin eksik eksikliği ve
RAG2) [genler RAG1 ve/veya RAG2 (11p13-p12),R]. Sayesinde
Bununla birlikte, bu enzimlerin düşük kalıntı aktivitesi nedeniyle, çoğaldıkları ve büyük miktarlarda IL-4 ve ürettikleri cilt ve sindirim sisteminin epitel dokularının antijenlerine özgü belirli sayıda T-lenfosit klonu gelişir. IL-5, hipereozinofiliye ve kalıntı B-lenfositleri tarafından IgE oluşumuna neden olur (diğer sınıfların immünoglobulinlerinin yokluğunda). Kafa derisi ve kaşlarda alopesi ile eritroderma ve pakidermi, zayıflatıcı ishal, hayatı tehdit eden bulaşıcı sendrom; hepatosplenomegali ve lenf nodu hiperplazisi.
. İyonlaştırıcı radyasyona karşı aşırı duyarlılığı olan SCID. Artemis nükleer protein defekti [gen DCLRE1C, (10p),R], DNA onarımı için gerekli enzimler kompleksine dahil edilir (çift iplikli kopmaların bağlantısına katılır), gen mutasyona uğradığında, V (D) J-rekombinasyonunun ihlali meydana gelir; T-B-NK+.
. IL-2 eksikliği [gen IL-2, 4q26-q27].
a zinciri IL-2 reseptör genindeki (CD25) mutasyonlar (10r15-r14);T - B + NK + .
IL-7 reseptörünün (CD127) a-zincir genindeki mutasyonlar (5r13);T - B + NK + .
TAP açığı (Antijen Sunumu için Taşıyıcı), IL-7 reseptörünün (CD127) a-zincir geninin endoplazmik retikulumuna antijenik peptitlerin taşınması için gereklidir. (5r13);T - B + NK + .
CD3 zincirlerinin (CD3γ, CDδ ve CDε) genlerindeki mutasyonlar, olgun T-lenfositlerin sayısında azalmaya, farklılaşmalarının ihlaline yol açar; T - B + NK + .
Protein tirozin kinaz ZAP-70 [gen eksikliği ZAP-70 (2q12), R]. Gen mutasyona uğradığında, TCR ζ zincirinin ITAM alanlarının ve ITAM içeren NK hücre reseptörlerinin fosforilasyonu zarar görür, CD8 + T hücrelerinin seçici bir eksikliği gelişir (CD4 + T-lenfositlerinin içeriği normaldir, ancak fonksiyonel bozukluklar bu hücreler tarafından IL- oluşumunun yokluğu şeklinde ifade edilir) 2 ve çoğalma).
Adenozin deaminaz eksikliği [gen ada (20q12-q13.11 , p)], hücrelerde metabolitlerin birikmesine yol açar (deoksiadenozin trifosfat ve S-adenosilhomosistein), bu da T- ve B-lenfositlerin proliferasyonunu inhibe eder (hastalığın geç başlangıçlı varyantları tarif edilmiştir); T-B-NK-.
Purin nükleosit fosforilaz eksikliği [gen pnp (14q11.2), p], hücrelerde T-lenfositlerin proliferasyonunu engelleyen deoksiguanozin trifosfat birikmesine yol açar (ilişkili sendromlar ürisemi ve ürikürdür); T - B + NK - .
Laboratuvar çalışmalarından elde edilen veriler. Değişken, bazen derin lenfopeni ortaya çıkarın; lenfositler, belirli bir antijene yanıt olarak çoğalamazlar; kan serumundaki immünoglobulin seviyesindeki bir azalma sıklıkla ifade edilir. Göğüs röntgeninde timusun gölgesi yok.
klinik tablo. Genellikle klinik tanı, maternal IgG antikorlarının kaybolduğu yaşamın ilk 6 ayında netleşir. Klinik tabloda öne çıkıyor şiddetli bulaşıcı sendrom lenfoid doku hipoplazisi ve gelişimsel gecikme. Enfeksiyöz sendrom, oral kandidiyaz, kronik ishal, pnömoni, ateş,
bakteriyel etiyoloji sepsisi, viral enfeksiyonlar. Enfeksiyöz ajanlar farklı taksonomik gruplara aittir: bakteriler, virüsler, mantarlar, fırsatçı mikroorganizmalar (Pneumocystis carini). Pnömoni sıklıkla neden olur P. carini, ishal - rotavirüsler, Campylobacter, Giardia lamblia. Genellikle viral hepatit gösterir. Bölgesel veya genelleştirilmiş BCG'nin gelişimi aşılamadan sonra karakteristiktir.
Tedavi parenteral beslenme, intravenöz immünoglobulinin tanıtımı, antibiyotiklerin atanması, antifungal ve antiviral ilaçlar dahil olmak üzere bakım tedavisinin atanmasını sağlar. İyileşmeyi sağlamak için ana tedavi yöntemlerinden biri, SCID'li çocukların genellikle yaşamın 1. yılında öldüğü kemik iliği naklidir. Bir çocuğun özellikle sterilize edilmiş koşullarda 2-3 yıla kadar yaşadığı izole vakalar tanımlanmaktadır. Yenidoğanlarda SCID'yi mümkün olduğunca erken tanımak önemlidir, çünkü onlar için örneğin canlı aşılarla bağışıklama ölümcüldür. Tanı konulduktan hemen sonra bu tür çocuklar gnotobiyolojik koşullara (steril kutu) yerleştirilmelidir. Bulaşıcı hastalıklara katılma durumunda yoğun antibakteriyel, antiviral ve antifungal tedavi, intravenöz immünoglobulin replasman tedavisi yapılır. Pnömokistik pnömoninin önlenmesi için ko-trimoksazol reçete edilir. BCGit'in gelişmesi durumunda, uzun süreli yoğun anti-tüberküloz tedavisinin yapılması gerekir. Kan bileşenlerinin transfüzyonu için sadece ışınlanmış ve filtrelenmiş müstahzarlar kullanılmalıdır. Maternal lenfositlerin transplasental transferi nedeniyle transfüzyon sonrası graft-versus-host hastalığı riski vardır.
Çıplak lenfosit sendromu
Bu, MHC-I veya MHC-II moleküllerinin vücutta eksprese edilmediği patolojinin adıdır. MHC-I moleküllerinin ekspresyonunun yokluğunda, CD8+ T-lenfositlerin içeriği azalır ve NK hücrelerinin aktivitesi yoktur; MHC-II'nin yokluğunda, CD4 + T-lenfositlerin seviyesi azalır. Çeşitli genetik kusurlar karakterize edilmiştir. Bununla birlikte, bu kusurlar MHC genlerinde değil, düzenlenmesinden sorumlu birkaç farklı faktörde lokalizedir.
ifade. Klinik tabloÇıplak lenfosit sendromu ve tedavi diğer SCID'ler için olanlara benzer.
DiGeorge Sendromu
DiGeorge sendromunda veya üçüncü ve dördüncü faringeal ceplerin kusur sendromunda [silmeler 22q11, gen dahil TBX1 (22q11.2), timus hipoplazisini veya aplazisini, paratiroid bezinin hipoplazisini, kalp kusurlarını, T-lenfosit eksikliğini, değişken miktarlarda B-lenfositlerini ortaya çıkarır.
Laboratuvar çalışmalarından elde edilen veriler. CD3+, CD4+ ve CD8+ T hücrelerinin sayısında önemli bir azalma ve mitojenler ve antijenler tarafından indüklenen proliferatif aktivitelerinde keskin bir azalma. B ve NK hücrelerinin sayısı normaldir. Çoğu durumda serum immünoglobulinlerinin konsantrasyonu normal aralıktadır, çeşitli disgamaglobulinemi varyantları mümkündür.
klinik tablo.İmmün yetmezlik bileşeni, timusun hipoplazisi veya aplazisi ve tekrarlayan, şiddetli bulaşıcı hastalıklar. Hipoparatiroidizm de tespit edilir (hipokalsemi ve bunun sonucunda doğumdan 1-2 gün sonra farkedilen tetani); dolaşım sisteminin malformasyonları (aort kemerinin sağ ters dönüşü, sağ ventrikül darlığı, interventriküler ve interatriyal septadaki kusurlar, Fallot tetralojisi, pulmoner arterin atrezi veya hipoplazisi); damak boşluğu; yüz iskeletinin anomalileri (eşleştirilmiş organlar arasındaki artan mesafe, çeneler, özellikle alt kısım, düşük kulak kepçeleri, kısa burun oluğu). Larinks, farinks, trakea, iç kulak, yemek borusu yapısında belirgin anomaliler; böbrek gelişimi, merkezi sinir sistemi ve diğer malformasyonlar (polidaktili, tırnak yokluğu, anüs atrezisi, anal fistüller). Konuşma ve psikomotor gelişimde bir gecikme karakteristiktir. yatkınlığı belirtmek otoimmün bozukluklar(sitopeni, otoimmün tiroidit) ve malign neoplazmalar.
Tedavi.. Antibakteriyel ve antiviral tedavi. . İntravenöz immünoglobulin preparatları ile değiştirme tedavisi. . Malformasyonları düzeltmek için cerrahi tedavi. . Otoimmün komplikasyonlarla - immünosupresif tedavi. . Endokrinopatilerin varlığında - ilgili ihlallerin düzeltilmesi. . Kemik iliği nakli etkisiz
tive. . Timus epitel doku nakli haklı. Paratiroid bezlerinin işlevinin düzeltilmesi.
X'e bağlı lenfoproliferatif sendrom
X- bağlantılı lenfoproliferatif sendrom, Epstein-Barr virüsüne [gendeki kusurlar nedeniyle] bozulmuş bağışıklık tepkisi ile karakterize edilir. SH2D1A(SAP) içinde xq25,אּ], Epstein-Barr virüsü tarafından dönüştürülen B-lenfositlerin kontrolsüz çoğalmasına ve virüs tarafından yeni hedef hücrelerin enfeksiyonuna yol açar.
klinik tablo. En yaygın 4 fenotip tanımlanmıştır: şiddetli enfeksiyöz mononükleoz, malign lenfoproliferatif durumlar (lenfomalar, lösemiler, ağırlıklı olarak B-hücresi), anemi veya pansitopeni (virüs kaynaklı hemofagositik sendrom nedeniyle dahil), disgamaglobulinemi. Epstein-Barr virüsü enfeksiyonu, en şiddetli, hızlı ilerleyen ve ölümcül hastalıkların oluşumu için bir tetikleyici (başlangıç) mekanizmadır: fulminan enfeksiyöz mononükleoz (vakaların %58'inde ölüme yol açar), hemofagositik sendrom (%100'ünde tedavi olmadan). ölüme neden olduğu durumlarda). Vakaların% 10'unda fenotip, Epstein-Barr virüsü ile enfeksiyondan önce ortaya çıkar (bu durumda, kural olarak, disgamaglobulinemi ve lenfomalar gelişir). En yaygın olanları çeşitli hipogamaglobulinemi türleridir. İmmün yetmezlik bakteri, mantar ve viral hastalıkların gelişmesine yol açar. bulaşıcı hastalıklar. Karakteristik aile öyküsü ve sero veya PCR pozitif Epstein-Barr virüs testi olan erkek çocuklarda hastalıktan şüphelenilebilir. Tanı için bir genetik analiz kombinasyonu önerilir. SH2D1A ve SAP ifade seviyesinin değerlendirilmesi.
Tedavi
Önleme amacıyla, antiviral ilaçlar - asiklovir, valasiklovir (erken uygulamaları Epstein-Barr virüsünün orofarenkste replikasyonunu baskılar) ve intravenöz immünoglobulin (Epstein-Barr virüsüne karşı yüksek titreli antikorlarla) kullanılması önerilir. ).
Hipogamaglobulinemi ile, antibiyotik tedavisi ile kombinasyon halinde aylık intravenöz immünoglobulin kullanılır.
Fulminan enfeksiyöz mononükleozun tedavisi için, yüksek dozlarda asiklovir ve metilprednizolon reçete edilir, Epstein-Barr virüsü ve IFN'ye karşı yüksek titre antikorlu intravenöz immünoglobulin ile yüksek doz tedavisi.
Hemofagositik sendromun gelişmesiyle birlikte yüksek dozlarda deksametazon vepezid ♠ (etoposid) ile birleştirilir.
Malign hastalıkların tedavisinde standart tedavi protokolleri kullanılmaktadır.
Radikal bir tedavi yöntemi, HLA uyumlu donörlerden kemik iliği naklidir.
Otoimmün lenfoproliferatif sendrom
Otoimmün lenfoproliferatif sendrom - iyi huylu lenfoproliferasyon, hiperimmünoglobulinemi, otoimmün bozukluklar, periferik kanda artan CD3 + CD4 - CD8 - T-lenfosit seviyeleri (çift negatif) ve apoptozda bir kusur [gen defekti] ile karakterize bir hastalık grubu Fas(CD95)- TNFRSF6 (10q24.1), gen kaspaz-10, Fas-ligandı - FasL(1q23)].
Laboratuvar çalışmalarından elde edilen veriler. Periferik kan veya lenfoid dokulardaki CD3 + CD4 - CD8 - T-lenfositlerin içeriği %1'den fazladır. IgG, IgA ve IgM seviyeleri normal, yükselmiş ve hatta azalmış olabilir. Yaşla birlikte, hipergamaglobulinemi, agamaglobulinemiye kadar düşük konsantrasyonda serum immünoglobulinleri ile değiştirilir. Eritrositlere, trombositlere, nötrofillere, düz kaslara, faktör VIII'e karşı otoantikorlar tespit edilir; antinükleer ve antifosfolipid otoantikorlarının yanı sıra romatoid faktör vb. Lenfositoz karakteristiktir.
klinik tablo. Tüm hastalarda karaciğer, lenf düğümleri (yaşamın ilk 5 yılında) ve dalak büyümüştür. Lenfoproliferasyona ateş ve gece terlemeleri eşlik etmez. ilk otoimmün reaksiyonlar lenfoproliferasyon ile çakışmayabilir ve daha sonra ortaya çıkabilir. Yaşla birlikte otoimmün reaksiyonların şiddeti artar. Daha sıklıkla kan hücrelerine (hemolitik anemi, trombositopeni, nötropeni) karşı otoimmün reaksiyonlar gelişir, daha az sıklıkla diğer organlar etkilenir. Malign neoplazmalar (T- ve B-lenfomalar, Burkitt lenfoma, atipik lenfoma, lenfogranülomatozis, vb.) gelişme riski artar.
Tedavi.. Kemoterapötik ajanlar (siklofosfamid, azatioprin, metotreksat, klorambusil). . Glukokortikoidler. . Şiddetli hipersplenizm ve hemositopeni ile splenektomi. . Ağır vakalarda kemik iliği nakli mümkündür.
Hiperimmunoglobulinemi sendromu E
Hiper-IgE sendromu, serum IgE seviyesinde önemli bir artış, cildin tekrarlanan apseleri ve stafilokok etiyolojisinin deri altı dokusu, pnömosel oluşumu ile pnömoni, yüz iskeletinin yapısındaki anomaliler, AD ile karakterizedir. Hiper-IgE sendromunun moleküler genetik yapısı henüz belirlenmemiştir. Bazı durumlarda otozomal dominant kalıtım, bazılarında ise otozomal çekinik kalıtım tanımlanmıştır. Kusurların sitokin reseptörlerinin sinyal moleküllerini etkilediği varsayılmaktadır (bu sendromun otozomal dominant formunda, durum3) ve muhtemelen Th17 hücre alt popülasyonunun bozulmuş işleyişi ile ilişkilidir. Hiper-IgE sendromunun oluşumundan sorumlu başka bir gen, kromozom 4 üzerinde lokalizedir. (4q).
Laboratuvar çalışmalarından elde edilen veriler.Çeşitli immünolojik bozukluklar tespit edilir: serumda IgE seviyesinde bir artış, nötrofil kemotaksisinin ihlali, antikor oluşumunda bir kusur; HRT'nin kandidin, difteri ve tetanoz toksoidlerine yanıtında azalma; yanıt olarak T hücrelerinin proliferatif aktivitesinin zayıflaması kandida ve mitojenlere tepkiyi sürdürürken tetanoz toksoidi. Periferik kanda ve cilt apselerinin sıvısında eozinofili. T ve B hücrelerinin sayısı normaldir.
klinik tablo. Erken yaşta orta derecede egzama. Yüzün karakteristik özellikleri (geniş burun köprüsü, geniş kalkık burun, yüz iskeletinin asimetrisi, çıkıntılı alın, derin gözler, yüksek damak). İskeletin gelişimindeki anomaliler, skolyoz, eklemlerin artan hareketliliği, küçük yaralanmalardan sonra kemik kırılma eğilimi ve diş değişiminin ihlali ortaya çıkar. Deri, deri altı doku ve lenf düğümlerinde apseler vardır. Pnömoni daha büyük yaşta gelişir (en yaygın patojenler S. aureus ve H. in-
grip), vakaların %77'sinde bir pnömosel oluşur, buna bağlı bir enfeksiyon P. aeruginosa ve A. fumigatus. Pnömoni ateş olmadan da ortaya çıkabilir. Mukoza zarlarının ve tırnakların kronik kandidiyazisi vakaların %83'ünde gelişir.
Tedavi.. Uzun süreli (önleme amaçlı - yaşam boyu) antibakteriyel ve antifungal tedavi. . Dermatit tedavisi için topikal ajanlar, ciddi vakalarda düşük dozlarda siklosporin A kullanılır. . Kemik iliği nakli etkisizdir.
KROMOZOM BOZUKLUĞU SENDROMLARI
Kromozomal kararsızlığı olan sendromlar için: ataksielanjiektazi[DNA topoizomeraz genindeki kusur ATM (11q22), p] ve Nijmegen sendromu[nibrin gen kusuru NBS1(8q21)] - artan malign tümör sıklığı, spontan kromozomal instabilite ve kromozomal bozulmalar karakteristiktir. Her iki protein de DNA çift zincir kırıklarının onarımında ve hücre döngüsünün düzenlenmesinde rol oynar. Normal olarak, DNA çift sarmal kopmaları, immünoglobulin ve TCR genlerinin V(D)J rekombinasyonu sırasında, immünoglobulin sınıflarını değiştirirken, çaprazlama sırasında ve mayoz sırasında meydana gelir. Benzer süreçler beyin nöronlarının olgunlaşması sırasında meydana gelir. Ataksi-telanjiektazi ve Nijmegen sendromunda DNA onarımındaki kusurlar, bozulmuş immünoglobulin sentezi, genital organların işlevleri ve sinir sistemi gibi klinik belirtilere neden olur.
ataksi-telanjiektazi
Çok heterojen bir fenotipe sahip bu sendrom (sıklık 1:300 bin yenidoğan), Fransız doktor D. Louis-Bar tarafından tanımlandı. Ataksi belirtileri zaten 2-4 aylık bir çocukta tespit edilebilir. Ataksi, beyincikteki Purkinje hücrelerinin ilerleyici dejenerasyonundan kaynaklanır. Burun derisinde, kulak kepçelerinde ve konjonktivada telenjiektaziler 3-6 yıl sonra ortaya çıkar. Café au lait lekeleri genellikle ciltte görülür. Timus, lenf düğümleri, dalak, bademciklerin hipoplazisi ile karakterizedir. İmmün yetmezlik, IgA, IgE, IgG2, IgG4 üretiminde bir azalma (genellikle bir dengesizlik) ile kendini gösterir. hastaların %80'i gelişir
karşılık gelen bulaşıcı klinik semptomlar. T hücrelerinin (esas olarak CD4 + T hücreleri) azaltılmış sayısı ve fonksiyonel aktivitesi. Çoğu hastada toplam T-lenfosit sayısı normaldir. Neoplazmaların (esas olarak lenfomalar ve karsinomlar) sıklığı olağandışı bir şekilde yüksektir (genel popülasyondan 200 kat daha fazla), genellikle 10-12 yaş arasında ölüme yol açar. Tedavi semptomatik.
Nijmegen sendromu
Nijmegen sendromu (adını, hastalığın ilk tanımlandığı Hollanda'daki şehirden alır) mikrosefali, yüz iskeletinin spesifik bozuklukları (eğimli alın, yüzün çıkıntılı orta kısmı, uzun burun, alt çene hipoplazisi, Moğol göz yarığı, epikantus) ile kendini gösterir. , büyük kulaklar), fiziksel gelişim gecikmesi , ciltte "sütlü kahve" lekelerinin varlığı; klinodaktili ve sindaktili, yumurtalık disgenezisi, vb. Çoğu çocuk tekrarlayan ve kronik bakteriyel enfeksiyondan muzdariptir. bulaşıcı hastalıklar solunum yolu, KBB organları ve üriner sistem. Vakaların %50'sinde, ağırlıklı olarak B hücreli lenfomalar olmak üzere malign neoplazmalar gelişir. CD4 + T hücrelerinde bir azalma olan çeşitli disgamaglobulinemi formlarını ortaya çıkarın.
Tedavi.. Nörolojik bozuklukların semptomatik tedavisi. . İntravenöz immünoglobulin ile değiştirme tedavisi. . Endikasyonlara göre antibakteriyel, antiviral, antifungal tedavi kullanılır. . Malign neoplazmların tedavisinde radyasyona ve kemoterapiye karşı artan hassasiyet dikkate alınır.
Wiskott-Aldrich Sendromu
Wiskott-Aldrich sendromu [kusurlu gen WASP (Xp11.23p11.22),אּ; ayrıca ρ ve Ʀ] Gen YABAN ARISI(itibaren Wiskott-Aldrich sendromu lenfositlerde, dalak dokusunda ve timositlerde eksprese edilir. Bu genin mutasyonları, CD43 molekülünün nötrofillerinde ve T-lenfositlerinde (CD4 ve CD8) anormal ekspresyonla ilişkilidir (ICAM-1 için ligand, yapışma önleyici bir işlev gerçekleştirir).
Laboratuvar çalışmalarından elde edilen veriler. Trombositopeni (normalin %10'undan azı) artan hücre yıkımından kaynaklanır.
Trombositler sağlıklı insanlara göre daha küçüktür. Kan serumundaki IgM seviyesi, normal bir IgG seviyesi ve IgA ve IgE içeriğindeki bir artış ile azalır. İzohemagglutininlerin titreleri azalır, pnömokok, streptokok, Escherichia coli, salmonella polisakkarit antijenlerine karşı antikor oluşumu ve ayrıca antiviral antikorlar bozulur. Erken yaşta, kural olarak, lenfosit sayısı normaldir; 6 yıl sonra, lenfopeni (1x109 / l'den az), normal düzeyde B ve NK hücreleri olan CD3 + ve CD4 + T hücrelerinde azalma saptanmış. Eozinofili ve posthemorajik anemi gelişimi mümkündür. T hücrelerinin mitojenlere ve antijenlere karşı proliferatif tepkisi azalır, HRT zayıflar. Dalakta germinal merkezlerin normal yapıları ve T hücre bölgeleri saptanmaz.
klinik tablo. Hastalık bir üçlü belirti ile karakterizedir: trombositopeni, egzama ve tekrarlayan bulaşıcı hastalıklar. Hemorajik sendrom, zaten yenidoğan döneminde erken kendini gösterir (peteşiyal döküntü, sefalohematomlar, göbek yarasından kanama, bağırsak kanaması). Egzama hastaların %80'inde erken yaşlardan itibaren kendini gösterir. Yaşla birlikte, bağışıklık yetmezliği belirtileri artar: üst solunum yollarının bakteriyel bulaşıcı hastalıkları, solunum sistemi, sindirim organları, cilt; yaygın veya genelleştirilmiş herpes enfeksiyonu (Herpes simpleks ve suçiçeği zoster) sitomegalovirüsün yanı sıra mantar (mukoza zarlarının kandidiyazisi), daha az sıklıkla fırsatçı bulaşıcı hastalıklar. Otoimmün hastalıklar (hemolitik anemi, nötropeni, artrit, kutanöz vaskülit, ülseratif kolit, serebral vaskülit, glomerülonefrit, otoimmün trombositopeni) hastaların %70'inde tespit edilir. 5 yaşından büyük hastaların sıklığı artmıştır. malign neoplazmalar(esas olarak lenfoid doku tümörleri).
Tedavi.. Allojenik kemik iliği veya kök hücre nakli (ameliyatın başarı oranı doku uyumlu bir donörden nakil ile %90'a, haploidentik nakil ile %50'ye ulaşır). . İntravenöz immünoglobulin ile değiştirme tedavisi. . Antibakteriyel, antifungal ve antiviral ilaçların profilaktik uygulaması. . Hemorajik sendromu azaltmak için splenektomi yapılır. . Otoimmün komplikasyonlar ile immünosupresif tedavi reçete edilir.
FAGOSİTOZ HATALARI
kronik granülomatöz hastalık
Kronik granülomatöz hastalık, fagositlerin fonksiyonel aktivitesinin (oksijen radikallerinin aktif formlarının oluşumu, hücre içi öldürme ve fagositozlu patojenlerin parçalanması), kalıcı bakteriyel ve fungal enfeksiyonlar ve granülomatöz inflamasyonun gelişimi ile karakterizedir. Kronik granülomatöz hastalık, çeşitli genetik kusurları olan bireylerde gelişir [vakaların %65'inde - hastalığın X'e bağlı varyantı: gen gp91-phox (Xp21.1),אּ; vakaların %35'inde - otozomal resesif: gen f47-phox (7q11.23),ρ; gen p67-phox (1q25),ρ; gen p22-phox (16q24), p], NADP-oksidaz sisteminde bozukluklara yol açar. Kısa ömürlü (birkaç saat) nötrofillerin ölümüyle, öldürülmemiş bakteriler iltihabın odağına “sızar”. Makrofajlar uzun ömürlü hücrelerdir ve öncülleri (monositler) artan sayıda odağa göç eder (bu da makrofaj oluşumuna yol açar). granülomlar), fagositoz mikroorganizmaları, ancak onları öldüremezler.
Laboratuvar çalışmalarından elde edilen veriler. Serum immünoglobulinlerinin normal değerleri ve lenfositlerin alt popülasyonları ile karakterize edilir. Testlerde değerlendirilen nötrofiller tarafından peroksit radikallerinin oluşumu (luminole bağlı kemilüminesans veya tetrazolyum mavisi indirgemesi), keskin bir şekilde azalır veya yoktur. Bulaşıcı hastalıkların arka planına karşı, lökositoz, nötrofili, artmış ESR, anemi, hipergamaglobulinemi karakteristiktir.
klinik tablo.Çoğu durumda hastalık, yaşamın ilk yılında kendini gösterir. bulaşıcı sendrom(hücre içi ve hücre dışı patojenlerle enfeksiyonlar) ve granülom oluşumu. En tipik olanları: akciğerlerde hasar (tekrarlayan pnömoni, hiler lenf düğümlerinde hasar, akciğer apseleri, pürülan plörezi), sindirim sistemi, cilt apseleri ve lenfadenit. En yaygın patojenler katalaz pozitif mikroorganizmalardır: S. aureus, Aspergillus spp., bağırsak gram negatif bakteriler (E. coli, Salmonella spp., Serratia marcescens), daha az sıklıkta - Burkholderia cepacia ve Nocardia farcinica. Hepatik ve subdiyafragmatik apseler, osteomiyelit, pararektal apseler ve sepsis gelişimi karakteristiktir.
En şiddetli, yaşamı tehdit eden enfeksiyöz komplikasyon, akciğerlerin ve diğer organların (yağ dokusu, beyin, kemikler, eklemler, endokard) yaygın bir lezyonu olarak ortaya çıkabilen aspergillozdur. BCG aşısından sonra kronik granülomatöz hastalığı olan hastalar sıklıkla bölgesel lenf düğümlerini içeren aşıyla ilişkili bir enfeksiyon geliştirir. Kronik granülomatöz hastalığı olan hastalarda mikobakteriyel lezyon hem pulmoner hem de ekstrapulmoner lokalizasyona sahip olabilir ve uzun süreli bir seyir gösterir. Kronik granülomatöz hastalığı olan hastalar için fiziksel gelişimde bir gecikme karakteristiktir.
Tedavi.. Antimikrobiyal tedavi: ko-trimoksazol ve antifungal ilaçların (itrakonazol, vb.) profilaktik sürekli kullanımı; bulaşıcı komplikasyonlar durumunda, kombine antibakteriyel tedavi, antifungal tedavi ile kombinasyon halinde parenteral olarak (hücre içine nüfuz eden 2-3 bakterisidal antibiyotik) gerçekleştirilir. Aspergilloz gelişmesiyle birlikte, uzun süreli amfoterisin B veya kaspofungin kullanımı belirtilir. Mikobakteriyel enfeksiyonda, geniş spektrumlu antibiyotikler ile anti-tüberküloz ilaçları ile uzun süreli spesifik tedavinin bir kombinasyonu kullanılır. . Cerrahi tedaviye genellikle postoperatif yaranın takviyesi ve yeni odakların oluşumu eşlik eder. Belki ultrason kontrolü altında apsenin delinme drenajı. . Antibiyotik tedavisinin etkisizliği ile şiddetli enfeksiyöz komplikasyonların tedavisi için granülositik kütle, yüksek dozlarda IFNy ve G-CSF kullanmak mümkündür. . Kemik iliği nakli veya uyumlu bir kardeşten kordon kanı nakli, bulaşıcı komplikasyonlar ve graft-versus-host hastalığından ölüm riskinin minimum olduğu erken yaşta başarılı olabilir.
Lökosit yapışmasındaki kusurlar
Bugüne kadar 3 lökosit yapışma kusuru tanımlanmıştır. hepsinde var
tekrarlayan ve kronik bakteriyel ve fungal enfeksiyonlarla karakterize otozomal resesif kalıtım. Tip I, lökositlerde CD11/CD18 ekspresyonunun olmaması veya azalması, nötrofil kemotaksisinin bozulması, lökositoz (25x109'dan fazla), göbek kordonunun geç düşmesi ve omfalit gelişimi, zayıf yara iyileşmesi ve patojenin vücuda nüfuz ettiği yerde irin oluşumunun olmaması.
Tedavi.. Antibakteriyel tedavi: bulaşıcı ataklar ve profilaktik. . Şiddetli vakalarda, HLA uyumlu bir donörden kemik iliği nakli reçete edilir.
TAMAMLAMA SİSTEMİNİN KUSURLARI
Kompleman Eksikliği Hastalıkları
Kompleman sisteminin tek tek bileşenlerindeki gen kusurlarının belirtileri Tablo'da verilmiştir. 11-2.
kalıtsal AO. Kompleman bileşenlerinin eksikliğinden kaynaklanan hastalıklar nadiren tespit edilir, çünkü tezahürleri otozomal aleller için homozigot bir durum gerektirir. C1inh (C1-esteraz inhibitörü) ile ilgili bir istisna vardır: bir gen mutasyonu c1inh, heterozigot durumda inhibitör eksikliğine yol açan, kalıtsal AO olarak bilinen bir fenotip ortaya çıkar (daha fazla ayrıntı için bkz. Bölüm 13, Anjiyoödem).
Bağışıklık komplekslerinin hastalıkları. C1-C4 eksikliği, toplu olarak sistemik lupus eritematozus (SLE) sendromu olarak adlandırılan sistemik vaskülit ve böbrek hasarı gibi bağışıklık kompleksi hastalıklarının gelişimi ile kendini gösterir.
piyojenik enfeksiyonlar. C3 eksikliği (ayrıca H ve I faktörleri), piyojenik enfeksiyonlara karşı artan duyarlılıkla ilişkilidir. Kompleman aktivasyonunun alternatif yolunda yer alan bileşenlerin eksikliği ve ayrıca C5-C8 bileşenlerinin eksikliği, aşağıdakilerin neden olduğu enfeksiyona karşı artan duyarlılıkla ilişkilidir. Neisseria spp. C9 eksikliği genellikle klinik olarak asemptomatiktir.
Tablo 11-2. Kompleman sisteminin bireysel bileşenlerindeki kusurların klinik belirtileri
Mannoz bağlayıcı lektin eksikliği
Mannoz bağlayıcı lektin eksikliği (mannoz bağlayıcı lektin - MBL olarak da adlandırılır) bir gen kusurundan kaynaklanır MBL(gende çeşitli nokta mutasyonları ve delesyonları) MBL Kafkasyalıların %17'sinde bulunur). Gen kusurları ile C2 ve C4 kompleman bileşenlerini parçalayan proteazların aktivasyonu ve lektin yolu boyunca kompleman sisteminin aktivasyonu bozulur. klinik olarak bu patoloji bulaşıcı bir sendromla kendini gösterir.
Laboratuvar çalışmalarından elde edilen veriler. Lenfositlerin, lökositlerin ve ayrıca immünoglobulin izotiplerinin alt popülasyonlarının analizi, klinik semptomlar için yeterli olan önemli sapmalar göstermez. Kan serumunda MSL yoktur.
Tedavi. Bu hastalık klasik bir immün yetmezlik değildir. Bu nedenle, immünotropik ajanlarla immüno-düzeltme kontrendikedir. Rekombinant MSL, bu kalıtsal kusuru olan hastalarda etyopatogenetik replasman tedavisi için farmakolojik bir ajan olarak kullanılabilir. Bu ilaç şu anda klinik deneylerden geçmektedir.
Çocuklarda geçici hipogamaglobulinemi
Çocuklarda geçici hipogamaglobulinemi, immünoglobulin sisteminin kademeli oluşumunun fizyolojik bir özelliği ile ilişkilidir. IgM ve IgA antikor oluşumunun olgunlaşması büyük ölçüde "geciktirilir". Sağlıklı çocuklarda maternal IgG içeriği giderek azalır ve altı ay sonra kendi IgG antikorlarının üretimi artar. Ancak bazı çocuklarda immünoglobulin seviyelerindeki artış gecikir. Bu tür çocuklar tekrarlayan bakteriyel enfeksiyonlardan muzdarip olabilir. Bu durumlarda, donör immünoglobulin preparatlarının infüzyonlarına (intravenöz immünoglobulin verilmesi) başvurulmamalıdır.
Seçici immünoglobulin A eksikliği
İmmünoglobulin A'nın seçici eksikliği (SD IgA - IgA'nın Seçici Eksikliği) bir gen kusurunun sonucu olarak gelişir tnfrsf13b
veya r). Diğer sınıfların immünoglobulinlerinin varlığında IgA eksikliği, genel popülasyonda 1:500-1500 kişi sıklığında (alerjisi olan hastalarda, hatta daha sık) tespit edilen en yaygın immün yetmezliktir. Seçici IgA eksikliği vardır, yani. alt sınıflardan birinin eksikliğinden (vakaların %30'u) ve tam (vakaların %70'i) oluşur. IgA2 alt sınıfının eksikliği, IgA1 alt sınıfının eksikliğinden daha belirgin bir klinik tabloya yol açar. IgA eksikliğinin diğer bozukluklarla kombinasyonları da mümkündür: IgG biyosentezinde bir kusur ve T-lenfosit anormallikleri ile. Seçici olan bireylerin büyük çoğunluğu
IgA eksikliği pratik olarak sağlıklıdır. 2 yaşın altındaki çocuklar için IgA eksikliği fizyolojik bir durumdur.
Serum IgA konsantrasyonunda bir düşüş tespit edin.<5 мг/дл у детей старше 4 лет; IgG и IgM в норме, количество и соотношение субпопуляций лимфоцитов и их функциональная активность могут быть в норме.
klinik tablo. IgA eksikliği ile 3 grup patolojik sendrom gelişebilir: bulaşıcı, otoimmün ve alerjik. IgA eksikliği olan hastalar, üst solunum yolu ve sindirim organlarının tekrarlayan bulaşıcı hastalıklarına yatkındır. En sık ve şiddetli olan çeşitli otoimmün hastalıklardır (romatoid artrit, ankilozan spondilit, Sjögren sendromu, beyin damarlarına zarar veren vaskülit, otoimmün tiroidit, SLE, glomerülonefrit, hemolitik anemi, tip I diyabetes mellitus, vitiligo, vb.). Çölyak hastalığı insidansı, normal IgA'lı çocuklarda 10 kat daha fazladır. En sık saptanan alerjik belirtiler inek sütü protein intoleransı, atopik dermatit (AtD), bronşiyal astımdır.
Tedavi. Asemptomatik vakalar herhangi bir özel tedavi gerektirmez; bulaşıcı, otoimmün ve alerjik hastalıkların klinik belirtilerinin varlığında, standartlara uygun olarak tedavi yapılır.
Alıcıda IgA'ya karşı antiizotipik antikorların oluşma ve bunların neden olduğu transfüzyon komplikasyonlarının gelişme olasılığı yüksek olduğundan, ne seçici ne de tam IgA eksikliği için donör immünoglobulinleri ile ikame tedavisi endike değildir.
Agammaglobulinemi B hücre eksikliği ile
X'e bağlı agamaglobulinemi (Bruton hastalığı) tüm agamaglobulinemi vakalarının %90'ını oluşturur. Kusurlu genin taşıyıcılarının erkek ve oğulları (אּ, ρ) hastalanır btk (Xq21.3-q22), B-lenfosit spesifik protein tirozin kinaz Btk'yi kodlayan (Bruton'un tirozin kinazı- Bruton'un tirozin kinazı). Kusurun bir sonucu olarak, hücre içi sinyal yollarının ihlali, ağır immünoglobulin zincirlerinin rekombinasyonu, farklılaşma vardır.
B-lenfositlerine pre-B hücrelerinin replikasyonu. B hücre eksikliği olan hastaların %10'unda agamaglobulinemi otozomal çekinik olarak kalıtılır. Pre-B hücre reseptörü, sitoplazmik B hücre adaptör proteini (BLNK) ve gen dahil olmak üzere şu ana kadar altı genetik kusur tanımlanmıştır. Lösin-Zengin Tekrar İçeren 8 (LRRC8).
Laboratuvar çalışmalarından elde edilen veriler. Periferik B-lenfositleri yoktur. Kemik iliği sitoplazmada μ-zincirli pre-B hücreleri içerir. T-lenfositlerin sayısı ve T-lenfositler için fonksiyonel testler normal olabilir. Kandaki IgM ve IgA tespit edilemez; IgG mevcut olabilir, ancak küçük miktarlarda (0.4-1.0 g/l). Kan grubu antijenlerine ve aşı antijenlerine (tetanoz, difteri toksinleri vb.) karşı antikor yoktur. Nötropeni gelişebilir. Lenfoid dokunun histolojik incelemesi: Lenfoid foliküllerde germinal (germ) merkezler ve plazma hücreleri yoktur.
klinik tablo. Aile öyküsü bilinmiyorsa ortalama 3,5 yaşında tanı belirgin hale gelir. Hastalık, lenfoid doku hipoplazisi, şiddetli pürülan enfeksiyonlar, üst (sinüzit, orta kulak iltihabı) ve alt (bronşit, zatürree) solunum yollarının bulaşıcı hastalıkları ile karakterizedir; olası gastroenterit, piyoderma, septik artrit (bakteriyel veya klamidyal), septisemi, menenjit, ensefalit, osteomiyelit. Solunum yolu hastalıklarının en yaygın etken maddeleri şunlardır: Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus, ishal bağırsak bakterileri veya giardia Giardia lamblia. Ayrıca agamaglobulinemili hastalar, kronik pnömoni, pürülan artrit, sistit ve deri altı doku apselerinin nedeni olan mikoplazmalar ve üreaplazmaların neden olduğu bulaşıcı hastalıklara karşı hassastır. Virüslerden, nörotropik virüsler ECHO-19 ve coxsackie tipiktir ve hem şiddetli akut hem de kronik ensefalite ve ensefalomiyelite neden olur. Enterovirüs enfeksiyonlarının belirtileri dermatomiyozit benzeri sendrom, ataksi, baş ağrıları ve davranış bozuklukları olabilir. Hasta çocuklarda, canlı çocuk felci aşısı ile aşılama sırasında, kural olarak, poliomyelit virüsünün mukoza zarlarından uzun süreli atılımı, ayrıca, restore edilmiş ve artan virülans ile (yani, çocuk koleksiyonunda -
sağlıklı çocuklarda aşılanmış bağışıklık yetersizliği olan bir çocukla temas sonucunda gerçek bir çocuk felci enfeksiyonu riski yoktur). Agammaglobulinemideki otoimmün bozukluklar, romatoid artrit, skleroderma benzeri sendrom, skleredema, ülseratif kolit, tip I diabetes mellitus (Th1 bağışıklık tepkisinin baskınlığından dolayı) ile temsil edilebilir.
Fiziksel inceleme. Fiziksel gelişimdeki gecikmeye, parmakların şekline (bavul şeklinde parmaklar), göğüs şeklindeki değişikliklere, alt solunum yolu hastalıklarının karakteristiğine, lenf düğümlerinin ve bademciklerin hipoplazisine dikkat edin.
Tedavi.
Yerine koyma tedavisi: Yaşam boyunca her 3-4 haftada bir intravenöz immünoglobulin preparatları uygulanır. İmmünoglobulin dozları, hastanın serumunda yaş normunun alt sınırıyla örtüşen konsantrasyonlarını oluşturacak şekilde seçilir.
Gen Tedavisinin Olanağının Tartışılması - Gen btk klonlanmıştır, ancak aşırı ekspresyonu hematopoietik dokunun malign transformasyonu ile ilişkilidir.
Kalıcı nötropeni durumunda büyüme faktörleri kullanılır. Otoimmün patoloji belirtileri ortaya çıktığında, monoklonal antikorların (infliximab, vb.) Yazılması mümkündür.
Ortak Değişken İmmün Yetmezlik
Yaygın değişken immün yetmezlik (CVID), antikor sentezi ve hücresel immünitede bir kusur ile karakterize edilen bir grup sendromdur. CVID için güvenilir bir tanı kriteri, aşağıdaki belirtilerden biriyle kombinasyon halinde her iki cinsiyette iki veya üç ana izotipin immünoglobulin içeriğinde önemli bir azalmadır:
2 yaş üzeri hastalığın başlangıcı;
İzohemaglütininlerin yokluğu ve/veya aşıya düşük yanıt;
Agammaglobulineminin diğer nedenlerinin dışlanması.
Bazı hastalarda CVID gelişiminin nedeni, B hücrelerinin olgunlaşma ve hayatta kalma süreçlerinde yer alan molekülleri kodlayan genlerdeki mutasyonlardır: BAFF-R (B-hücresi Aktivasyon Faktörü Reseptörü), keşif balonu-1 (B-lenfosit kaynaklı olgunlaşma proteini-1) ve ICOS (İndüklenebilir kostimülatör). B-lenfositlerin plazma hücrelerine farklılaşma yeteneğinin ihlali vardır, antikor oluşumunda kusurlar gelişir, T-lenfositlerin işlevsizliği mümkündür ve bulaşıcı hastalıklara karşı artan bir duyarlılık gözlenir. Sendrom erken çocukluk, ergenlik veya genç erişkinlerde ortaya çıkabilir.
Laboratuvar çalışmalarından elde edilen veriler. IgG ve IgA'nın (hastaların yaklaşık %50'sinde) ve IgM'nin (saptanamayan miktarlara kadar) önemli ölçüde azalması. Kandaki B-lenfosit sayısı normaldir veya azalmıştır. Çoğu hastada T-lenfosit sayısı normaldir. Şiddetli hastalarda lenfopeni gelişebilir (1 litre kanda 1500x103 hücreden az). NK hücrelerinin sayısı azalır. Bağışıklamaya yanıt olarak spesifik antikorların üretimi azalır veya yoktur. Mitojenlerin ve antijenlerin etkisi altında lenfositlerin proliferasyonu ve IL-2 oluşumu önemli ölçüde bozulur.
klinik tablo. Tekrarlayan bakteriyel enfeksiyon hastalıkları, esas olarak solunum yolu ve paranazal sinüslerde lokalizasyon ile tespit edilir. Tanı anında solunum yolu enfeksiyonları bronşektazi ve yaygın akciğer dokusu lezyonlarına kadar ilerleyebilir. Belki de ishal, steatore ve malabsorpsiyon (ve buna bağlı olarak kilo kaybı) ile kendini gösteren sindirim sisteminin bulaşıcı bir lezyonu. Genellikle enfeksiyonlar neden olur Giardia lamblia, Pneumocystis carinii veya ailenin virüsleri Herpetoviridae. CVID'li hastalar, mikoplazmalar ve üreaplazmaların neden olduğu pürülan artrit gelişimine eğilimlidir. Ensefalomiyelit, çocuk felci ve dermatomiyozit benzeri sendromlar, deri ve müköz membran lezyonları enterovirüs enfeksiyonlarının belirtileri olabilir. otoimmün hastalıklar şiddetlidir ve CVID'nin prognozunu belirleyebilir. Bazen CVID'nin ilk klinik belirtileri artrit, ülseratif kolit ve Crohn hastalığı, sklerozan kolanjit, malabsorpsiyon, SLE, nefrit, miyozit, lenfoid interstisyel pnömoni, nötropeni şeklinde otoimmün akciğer hasarı,
trombositopenik purpura, hemolitik anemi, pernisiyöz anemi, total alopesi, retinal vaskülit, ışığa duyarlılık. CVID'li hastalarda önemli ölçüde artmış bir sıklık vardır. malign neoplazmalar(vakaların %15'inde), sarkoidoz benzeri granülomlar ve malign olmayan lenfoproliferasyon. Tedavi.
Antibakteriyel kemoterapi.
Yerine koyma tedavisi: Yaşam boyunca her 3-4 haftada bir intravenöz immünoglobulin preparatları uygulanır.
Otoimmün komplikasyonlarla - immünosupresif tedavi (glukokortikoidler, azatioprin, siklosporin A) ve monoklonal antikorların (infliximab, vb.) atanması mümkündür.
Hiper-IgM sendromları
Hiper-IgM sendromları, IgG, IgA ve normal veya yüksek serum IgM konsantrasyonlarında belirgin bir azalma veya tam yokluk ile karakterize oldukça nadir görülen hastalıklardır. Bunun nedeni, B-lenfositlerinin immünoglobulin sınıf geçişi ve değişken alan hipermutajenezi gerçekleştirememesidir. Bugüne kadar, hiper-IgM sendromunun gelişmesine yol açan 6 genetik kusur tanımlanmıştır.
. Tip 1 (HIGM 1). CD40 ligandının X'e bağlı eksikliği (hiper-IgM sendromlarının vakalarının %70'i), T hücrelerinin B lenfositleri ile etkili bir şekilde etkileşime girememesiyle sonuçlanır.
. Tip 2 (HIGM 2). Otozomal resesif, sitidin deaminazın (gen Aicda, 12p13)- immünoglobulinler ve hipermutajenez sınıfları arasında geçiş yapan bir enzim.
. Tip 3 (HIGM 3). Otozomal resesif, CD40 molekül genindeki bir mutasyonla ilişkili. Aynı zamanda, B-hücrelerinin kendileri, T-lenfositleri ile etkili bir şekilde etkileşime giremezler. Fenotipik belirtiler tip 1'dekine benzer.
. Tip 4 (HIGM 4). Otozomal çekinik; bazı durumlarda mutasyonlar meydana gelir de novo. UNG - urasil-DNA glikosilazdaki bir kusurla ilişkili - aynı zamanda dahil olan bir enzim
immünoglobulin sınıflarını değiştirirken, ancak AID'nin etkisinden sonra. Bu durumda hipermutajenez etkilenmez ve sendrom daha az şiddetlidir.
. Tip 5 (HIGM 5). Kusur sadece sınıf geçişindedir, hipermutajenez etkilenmez. Nedensel mutasyon henüz tanımlanmadı, ancak sonrasında etki eden enzimde açıkça bir kusur var.
. Tip 6 (HIGM-ED). Dishidrotik ektodermal displazi ile ilişkili X'e bağlı, CD40'tan bozulmuş sinyalleşmeye yol açan NEMO (NF-kB modülatörü) eksikliğinden kaynaklanır.
X'e bağlı hiper-IgM sendromu diğerlerinden daha sık tespit edilir. CD40L'yi kodlayan gende (CD154, genin bulunduğu gende) bir kusur ile gelişir. Xq26-q27.2)- CD40 için ligand. T-lenfositler tarafından CD40L ekspresyonunun yetersizliği, B-lenfositlerdeki immünoglobulin sınıflarının IgM'den diğer izotiplere geçişinin imkansızlığının yanı sıra, hafıza B hücrelerinin, T-hücre repertuarının ve yönlendirilmiş Thl-hücre yanıtının bozulmuş oluşumuna yol açar. hücre içi mikroorganizmalara karşı çocuklar hasta olsun
Laboratuvar çalışmalarından elde edilen veriler. IgG, IgA, IgE belirlenemez veya çok küçük miktarlarda tespit edilir. IgM seviyesi normaldir (vakaların %50'sinde) veya genellikle önemli ölçüde yükselmiştir. T ve B hücrelerinin sayısı normaldir; antijenler tarafından indüklenen T hücrelerinin azaltılmış proliferatif tepkisi. IgM poliklonal, bazen monoklonaldir. IgM izotipinin otoantikorları tespit edilir (antieritrosit, antiplatelet, antitiroid, düz kas dokusu antijenlerine karşı antikorlar). Lenfoid dokuda germinal merkez yoktur, ancak plazma hücreleri vardır.
klinik tablo.İlk belirtiler bebeklik ve erken çocukluk döneminde ortaya çıkar. Tekrarlanan ile karakterize enfeksiyonlar fırsatçı (nedeniyle oluşan) dahil olmak üzere farklı lokalizasyon (öncelikle solunum yolu) Pneumocystis carini). Virüs enfeksiyonları (sitomegalovirüs ve adenovirüsler) de karakteristiktir, Criptococcus neoformans, mikoplazmalar ve mikobakteriler. Cryptosporidial enfeksiyon, akut ve kronik ishale (hastaların %50'sinde gelişir) ve sklerozan kolanjite neden olabilir. Genellikle anemi, nötropeni, oral mukoza ülserasyonu, diş eti iltihabı, ülseratif gelişir.
yemek borusu lezyonları, bağırsağın çeşitli kısımları, ülseratif kolit. yatkınlık gösterir otoimmün bozukluklar(seronegatif artrit, glomerülonefrit, vb.) ve malign neoplazmalar (esas olarak lenfoid doku, karaciğer ve safra yolları). Lenfadenopati, hepato ve splenomegali gelişebilir. Tedavi
İntravenöz immünoglobulin ile düzenli replasman tedavisi.
Antibakteriyel kemoterapi. Pneumocystis pnömonisinin önlenmesi ve tedavisi için ko-trimoksazol [sülfametoksazol + trimetoprim] ve pentamidin kullanılır.
Karaciğer ve safra yollarına zarar vermemek için sadece kaynamış veya filtrelenmiş su kullanmalı, düzenli muayeneler (ultrason, endikasyon varsa karaciğer biyopsisi) yapmalısınız.
Nötropeni ve ağız boşluğunun ülserasyonunun tedavisinde glukokortikoidler ve granülosit koloni uyarıcı faktör müstahzarları kullanılır.
Otoimmün komplikasyonların gelişmesiyle birlikte, immünosupresif tedavi (glukokortikoidler, azatioprin, siklosporin A) ve monoklonal antikorlara dayalı ilaçlar reçete edilir.
Optimal tedavi, HLA uyumlu donörlerden kemik iliği naklidir (hayatta kalma oranı %68, en iyi 8 yaşından önce yapılır).
En sık görülen birincil immün yetmezliktir. 1/300 - 1/700 sıklıkta oluşur. İlk kez oldu…
1960 yılında J. Heremans tarafından tanımlanmıştır.
Çoğu durumda, hastalığın kalıtımının net bir resmi gözlenmez, daha nadir ailesel IgA kaybı vakalarında, otozomal resesif kalıtım baskın olandan daha yaygındır. IgA eksikliği bazen kromozom 18 anormallikleri ile ilişkilidir.
IgA, karbonhidrat ve sialik asit içeriği ve dimerler, trimerler ve hatta tetramerler oluşturma kabiliyeti bakımından diğer immünoglobulinlerden farklıdır. IgA moleküllerinin 2 çeşidi vardır:
- serum IgA - her zaman bir monomer;
- salgı IgA - sırların (tükürük, gözyaşı, süt, sindirim, solunum ve idrar yollarının sırlarının) ayrılmaz bir parçası olmadan önce di-, tri- ve tetramerler oluşturur.
Özel bir protein zinciri olan J zinciri, salgı IgA'nın oluşumunda yer alır. Bu nedenle, bütün bir protein sistemidir. Dolayısıyla IgA patolojisinin formları:
- IgA'nın genel eksikliği, IgA monomerinin sentezindeki anormallikler ile ilişkilidir. Sonuç olarak, hem serum hem de salgı IgA içeriği azalır. Hem yerel hem de genel koruma ihlal ediliyor;
- salgı moleküllerinin oluşumundaki kusur - SIgA. Bunun nedeni, yerel bağışıklığın ihlaline yol açan bir J zincirinin olmaması olabilir;
- sadece IgA'nın salgı formları salgılandığında ve serum IgA monomerleri pratik olarak salgılanmadığında, serum IgA sentezinin baskın bir ihlali.
Klinik semptomlar şunlara bağlıdır: IgA eksikliği derecesi ve bireysel organ ve sistemlerin patolojisi şeklinde kendini gösterebilir. Çoğu zaman, mukoza zarının enfeksiyöz lezyonları meydana gelir.
Gastrointestinal sistemden - bu kronik gastrit, ülseratif ve hemorajik kolit, ileit, stomatit. Solunum sistemi kısmında - rinit, sinüzit, bronşit, hızla kronikleşir. Genellikle bronşektazi ve akciğer apsesi ile sonuçlanan kronik bronkopnömoni vardır. Düşük salgı IgA içeriği ile artan atopik alerji insidansı ve otoimmün hastalıkların (sistemik lupus eritematozus, romatoid artrit, otoimmün tiroidit) gelişimi arasında da bir bağlantı bulundu.
Ağırlıklı olarak serum monomerik IgA'nın seçici eksikliği olan kişilerde, çoğu zaman immün yetmezliğin klinik belirtileri görülmez.
Laboratuvar araştırması: bu tür hastaların periferik kanındaki toplam T- ve B-lenfosit sayısının normal aralıkta olduğunu gösterir. IgM ve IgG içeriği normal sınırlar içindeyken, serum ve/veya salgı IgA seviyeleri keskin bir şekilde azalır. Bazı hastalarda (atopik alerji belirtileri olan) IgE seviyeleri yükselebilir.
Tedavi: semptomatik. İmmünoglobulin replasman tedavisini kullanırken dikkatli olmak gerekir, çünkü tekrarlanan uygulama ile IgG ve IgM sınıflarının anti-immünoglobulin üretiminin artması olasılığı nedeniyle anafilaktik şok riski vardır.
Tahmin etmek: Hastalığın seyri çoğunlukla iyi huyludur.
tanı kriteri- 4 yaşından büyük hastalarda, normal IgG ve IgM seviyeleri ile serum IgA seviyeleri 0.07 g / l'den az olan hastalarda azalma ve diğer hipogamaglobulinemi nedenlerinin dışlanması. Teşhis açısından önemli:
1 yaşından büyük çocuklarda normal bir immünoglobulin izotipi seviyesi ile serumda IgA seviyesinde (0.05 g / l'den az) izole bir azalma, IgAl ve IgA2 yokluğu. IgM ve IgG seviyeleri normaldir. Ancak bazı hastalarda IgG2 eksikliği vardır;
- IgA seviyesi 0,05 g/l ila 0,2 g/l aralığındaysa, kısmi IgA eksikliği teşhisi konulur; normal sayıda T-lenfosit ve alt sınıfları;
- genellikle normal sayıda B lenfosit (CD19\CD20);
- normal sayıda NK hücresi (CD16 CD56).
IgA eksikliği olan hastalardaözellikle sekretuar IgA'nın yokluğunda, IgA alt sınıflarının düzeyini araştırmak gerekir. Bazı hastalarda seçici IgA eksikliği CVID gelişimi ile ilerleyebilir. İmmünoglobulin içeriğinin uzun süreli düzenli olarak izlenmesi gereklidir (asemptomatik hastalar dahil).
Otoantikorların belirlenmesi (antinükleer, antitiroid vb.).
Yemekle hoşgörüsüzlük veya malabsorpsiyon, alerji testi ve süte karşı antikorların ve anti-gluten IgG antikorlarının belirlenmesi gereklidir.
Tedavi. Asemptomatik selektif IgA eksikliği olan hastalarda kalıcı tedavi gerekmez. Bulaşıcı hastalık belirtileri olan hastalara profilaktik amaçlar için antibiyotik reçete edilir. Bulaşıcı bir hastalığın başlangıcında tüm hastalarda yoğun antibiyotik tedavisi yapılır. Hastalar rutin bağışıklamada kontrendike değildir. Bir hastada anti-IgA otoantikorları tespit edildiğinde immünoglobulin replasman tedavisi kontrendikedir. Selektif IgA eksikliğinin, düzeltilmemiş primer immün defektleri ifade ettiği dikkate alınmalıdır. Terapötik önlemler, bulaşıcı, alerjik ve otoimmün hastalıkların semptomatik tedavisine indirgenmiştir. İmmünotropik ilaçlar, esas olarak artan bulaşıcı morbiditenin tezahürü ile bağlantılı olarak reçete edilir.
Tahmin etmek. Selektif IgA eksikliği olan hastalarda prognoz, spesifik antikorlarda, alerjilerde veya otoimmün hastalıklarda eşlik eden bir kusurun varlığına bağlıdır. Çoğu zaman, hastalığın asemptomatik seyri, örneğin stresli bir durumda, immünosupresyon, kemoterapi vb. Gibi dış zararlı faktörlerin etkisi nedeniyle bozulabilir.

