Selektivni nedostatak IgA najčešći je primarni poremećaj imunodeficijencije (PIDS). Incidencija bolesnika s selektivnim nedostatkom IgA kreće se od 1: 400 do 1: 1000 u bijeloj populaciji i mnogo niža, od 1: 4000 do 1: 20 000, u mongoloidima. U Sjedinjenim Državama, prevalencija bolesti kreće se od 1 na 223-1000 u ispitivanoj skupini do 1 na 400-3000 u zdravih darivatelja krvi. Takva istraživanja nisu provedena u Rusiji.
Ovo stanje karakterizira selektivno smanjenje koncentracije IgA u serumu ispod 0,05 g / l (u djece starije od četiri godine) s normalna razina ostali serumski imunoglobulini, normalna reakcija serumska antitijela i normalan imunološki odgovor posredovan stanicama. U većini studija incidencija među muškarcima i ženama bila je približno ista.
Osobe koje nisu u stanju proizvesti IgA mogu bolest nositi asimptomatski putem mehanizama kompenzacije ili pate od čestih respiratornih, probavnih ili genitourinarni sustav, gastroenterološka patologija (na primjer, celijakija), sklonost atopijskim poremećajima poput peludne groznice, bronhijalna astma, atopijski dermatitis, IgE-posredovano alergija na hranu, kao i neurološke i autoimune bolesti (najčešće je to reumatoidni artritis, sistemski eritemski lupus, idiopatska trombocitopenična purpura, Sjogrenov sindrom). S selektivnim nedostatkom IgA alergijske bolesti, poput atopijskog dermatitisa i bronhijalne astme, dogodili su se u 40% slučajeva (Consilium Medicum, 2006). Anafilaktičke reakcije tijekom transfuzije komponenata krvi i primjene intravenskih imunoglobulina također su tipične za većinu ovih pacijenata, što je povezano s prisutnošću IgA u tim proizvodima.
Klinički simptomi selektivnog nedostatka IgA mogu se očitovati u ranom djetinjstvu, dok se učestalost i težina prenesenih infekcija mogu smanjivati \u200b\u200bs godinama zbog kompenzacijskog povećanja antitijela iz podrazreda IgG1 i G3, IgM. Još jedno objašnjenje izostanka klinički simptomi može postojati normalna razina sekretornog IgA unatoč smanjenju razine serumskog imunoglobulina. Ili, naprotiv, neki pacijenti s početno dijagnosticiranim selektivnim nedostatkom IgA mogu razviti kliničku sliku uobičajenog varijabilnog imunološkog nedostatka.
Terapija selektivnog nedostatka IgA trenutno se sastoji u identificiranju popratnih bolesti, provođenju preventivne mjereopijum kako bi se smanjio rizik od infekcije, kao i za brzo i učinkovito liječenje infekcija.
Ne postoji specifičan tretman. Prognoza u bolesnika s nedostatkom IgA općenito je dobra, ako nema izraženih kliničke manifestacije... Nedostatak IgA u djece može se vremenom nadoknaditi.
Budući da su genetski određena, stanja imunodeficijencije proizlaze iz oštećenja genetskog aparata. Pacijenti s uobičajenim varijabilnim imunološkim nedostatkom i oni sa selektivnim nedostatkom IgA često se nalaze u istoj obitelji i imaju zajednički HLA haplotip; mnogi imaju rijetke alele i delecije gena unutar klase MCH - klase 3 na kromosomu 6. Nedavno je pokazano da su neki obiteljski slučajevi uobičajene varijabilne imunološke manjkavosti i selektivnog nedostatka IgA uzrokovani mutacijama u genu TNFRSF13B, koji kodira protein poznat kao TACI (transmembranski aktivator i modulator kalcija i interakcija ciklofilin-ligand). Vjerojatno je da u slučajevima kada TACI mutacije nisu pronađene, uzrok bolesti mogu biti spontane ili nasljedne mutacije drugih gena, koje još nisu zabilježene.
Trenutno su dovoljno detaljno opisane moguće kliničke manifestacije selektivnog nedostatka IgA, varijante tečaja i moguće popratne bolesti. Odlučujuće za dijagnozu bolesti je selektivno smanjenje koncentracije IgA u serumu u djece ispod 4 godine ispod 0,05 g / l s normalnim razinama ostalih serumskih imunoglobulina u ponovljenim imunogramima. Liječenje se sastoji u prepoznavanju popratnih bolesti, poduzimanju preventivnih mjera kako bi se smanjio rizik od infekcije, a također je potrebno brzo i učinkovito liječenje zarazne bolesti.
Nema podataka o učestalosti pojavljivanja ovog primarnog stanja imunodeficijencije u ruskoj populaciji, što onemogućava usporedbu prevalencije bolesti u našoj zemlji s drugim zemljama u kojima su slična istraživanja već provedena.
Glavni problem je nedostatak jedinstvenih preporuka o taktikama liječenja bolesnika s selektivnim nedostatkom IgA.
Kako bi se procijenila učestalost pojave selektivnog nedostatka IgA među djecom iz skupine dispanzersko promatranje "Često bolesna djeca" i opišite raspon njegovih kliničkih manifestacija u Ruska Federacija na temelju Savezne državne proračunske institucije "FNKTS DGOI imenovane po Dmitriju Rogačovu" Ministarstva zdravlja Ruske Federacije i GBUZ DGKB br. 9 po G.N.Speransky DZM ovaj je posao izveden.
Materijali i metode istraživanja
Predmet istraživanja bila su djeca sa selektivnim nedostatkom IgA, koja su uočena u GBUZ DGKB № 9 nazvanom po G.N.Speransky DZM. Pored toga, provedena je retrospektivna analiza medicinskih kartona za razdoblje od 2003. do 2010. godine. 9154 pacijenta iz skupine dispanzerskog promatranja "često bolesne djece" (tablice 1-3).
Tijekom pregleda korištene su sljedeće metode:
- klinički i anamnestički;
- općenito i biokemijske analize krv;
- imunološko proučavanje sastava krvi nefelometrijom i metodama protočne citometrije;
- testovi skarifikacije;
- određivanje specifičnog IgE imunoblotiranjem;
- proučavanje funkcije vanjskog disanja;
- rinocitološki pregled.
Dijagnoza selektivnog nedostatka IgA postavljena je na temelju selektivnog smanjenja koncentracije IgA u serumu ispod 0,05 g / L s normalne performanse drugi imunoglobulini u serumu u ponovljenim imunogramima i isključenje ostalih mogući razlozi njihov nedostatak kod djece starije od 4 godine.
Prilikom uzimanja anamneze posebna pažnja dana učestalost i spektar kliničkih manifestacija, popratna patologijaa detaljno se proučavala obiteljska povijest. Klinički pregled djece proveden je u skladu s općeprihvaćenim metodama. Sadržaj imunoglobulina klasa A, G, M, E u serumu određen je nefelometrijom na BN 100 nefelometru (Dade Bering, Njemačka) pomoću Dade Behring kompleta. Fenotipizacija limfocita provedena je protočnom citometrijom na uređaju FacsScan (Becton Dickenson, USA), koristeći fluorescentno obilježena monoklonska antitijela Simultest (Becton Dickenson, USA). Pacijenti s bilo kojim manifestacijama atopije, kao i svi bolesnici s povišena razina IgE, koji je otkriven kao rezultat procjene pokazatelja imunološkog statusa nefelometrijom, proveden je dodatnim alergijskim pregledom testovima skarifikacije u djece starije od 4 godine ili metodom određivanja specifičnog IgE u krvnom serumu u bolesnika mlađih od 4 godine. Djeca s utvrđenom dijagnozom bronhijalne astme ili anamnezom bronho-opstruktivnog sindroma podvrgnuta su proučavanju funkcije vanjskog disanja pomoću aparata Spirovit SP-1 (Schiller AG, Švicarska). Također su obavljena sva potrebna dodatna ispitivanja i konzultacije srodnih stručnjaka, uzimajući u obzir postojeće pritužbe.
Rezultati i njihova rasprava
Retrospektivna analiza medicinske dokumentacije bolesnika s preporučenim dijagnozama „recidivirajućeg ARVI-a“, „CWD-a“, „CBS-a“, kao i „EBD-a“ omogućila je utvrđivanje da je učestalost selektivnog nedostatka IgA u ovoj skupini djece dva ili čak tri puta veća od u populaciji.
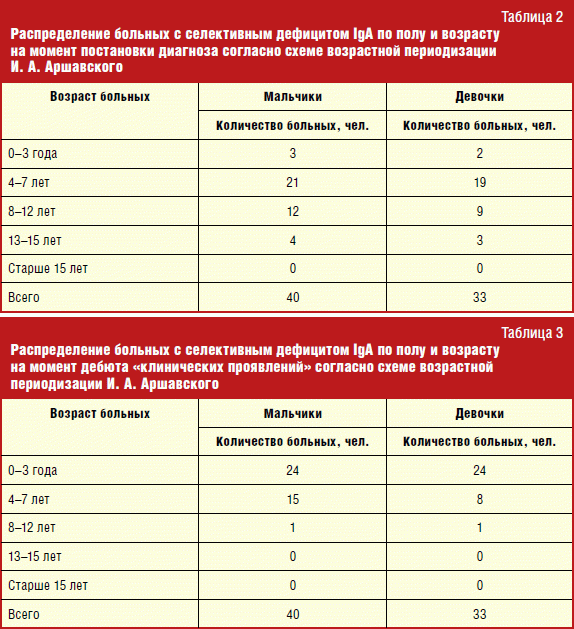
Apsolutni broj, kao i postotak djece s ovom primarnom imunodeficijencijom po godinama, može se vidjeti u tablici. 4.
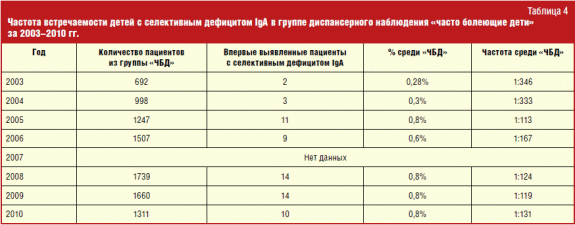
Nažalost, podaci za 2007. nisu dostupni. 2003. i 2004. god. Konzultirano je 692 i 998 djece. Među njima je identificirano ukupno 5 bolesnika sa selektivnim nedostatkom IgA, što je nešto češće od prosjeka populacije - 1: 346, odnosno 1: 333, u odnosu na 1: 400-600. Od 2005. godine učestalost novodijagnosticiranih pacijenata s ovim PIDS-om naglo je porasla: 1: 113 u 2005., 1: 167 u 2006., 1: 124 u 2008., 1: 119 u 2009. i konačno , 1: 131 u 2010. Tijekom istraživanja učestalost pojavljivanja promijenila se s 1: 346 u 2003. na 1: 131 u 2010., kada se pokazalo da je najviša u usporedbi s prethodnim godinama. Povećanje incidencije bolesnika s selektivnim nedostatkom IgA u trećoj godini nakon početka rada trebalo bi povezati s povećanom budnošću liječnika u odnosu na ovu patologiju, kao i s poboljšanjem laboratorijska dijagnostika... Potrebno je i dalje širiti znanje liječnika o ovoj bolesti, jer protok djece čiji roditelji dovode imunologu s pritužbama na česte bolesti, povećava se iz godine u godinu.
Tijekom ovog rada također je prospektivno pregledano 235 djece i 32 odrasle osobe.
Glavnu skupinu činilo je 73 djece s dijagnozom selektivnog nedostatka IgA.
Druga skupina bolesnika obuhvaćala je 153 djece s idiopatskom trombocitopeničnom purpurom (ITP). Procjena imunološkog statusa bolesnika s ITP-om provedena je kako bi se među njima identificirao selektivni nedostatak IgA, budući da je ta korelacija opisana u svjetskoj literaturi, a isti podaci dobiveni su tijekom ova studija... Među njima nismo pronašli niti jedno dijete s odsutnošću IgA. Unatoč činjenici da prilikom ispitivanja imunološkog statusa bolesnika s ITP nismo uspjeli među njima identificirati selektivni nedostatak IgA, utvrđeni su i drugi manji humoralni nedostaci: nedostatak podrazreda IgG, hipogamaglobulinemija dojenčadi, djelomično smanjenje IgA.
Treća skupina obuhvaćala je 32 odrasle osobe u dobi od 20 do 54 godine, kao i 8 djece u dobi od 4 do 10 godina, koja su najbliža rodbina bolesnika s selektivnim nedostatkom IgA, koji su prošli procjenu imunološkog statusa kako bi se pronašli i opisali obiteljski slučajevi.
Tijekom istraživanja i analize dobivenih podataka dobiveni su sljedeći rezultati.
Omjer muškaraca i žena među pacijentima s selektivnim nedostatkom IgA bio je približno jednak. Pregledano je 40 dječaka i 33 djevojčice. To odgovara podacima svjetske literature.
Vrhunac otkrivenosti selektivnog nedostatka IgA bio je u dobi od 4-7 godina. Ponovljene zarazne bolesti u pravilu su se dogodile u rano doba ili s početkom posjeta predškolskoj ustanovi. U pravilu su djeca prije nego što su došla do imunologa nagomilala određenu zaraznu povijest, jer postoje određeni znakovi koji im omogućuju sumnju na PIDS. I, uz to, čak i ako je studija provedena u ranijoj dobi i ako je otkrivena odsutnost IgA do 4 godine, to nam nije omogućilo postavljanje jednoznačne dijagnoze PIDS-a, ne bismo mogli u potpunosti isključiti nezrelost sustava sinteze imunoglobulina. Stoga je do starosti od 4 godine dijagnoza postavljena na temelju pitanja i preporučeno je praćenje. Stoga je interval od 4-7 godina.
Vodeće pritužbe u djece s selektivnim nedostatkom IgA bile su česte respiratorne bolesti virusne infekcije s nekompliciranim tečajem. Debi ponavljajućih respiratornih bolesti u pravilu je pao u dobi od 3 godine. To također odgovara podacima svjetske literature. Budući da se dinamička kontrola nad većinom pacijenata naše studije provodila dulje vrijeme, nekoliko godina, ponekad i prije prelaska pacijenta na mrežu odraslih, može se tvrditi da su se učestalost i težina prenesenih infekcija smanjivale s godinama. Vjerojatno je to bilo zbog kompenzacijskog povećanja antitijela iz podrazreda IgG1 i IgG3, IgM, ali ovo pitanje zahtijeva daljnje proučavanje. Druga najčešća pritužba tijekom liječenja bile su česte akutne respiratorne virusne infekcije s komplikacijama. Učestalost kompliciranih, atipičnih ARVI s godinama u naših se pacijenata, kao što je pokazano dinamičkim promatranjem, također smanjila.
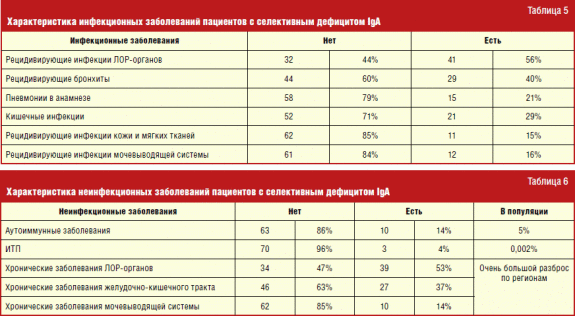
Među spektrom zaraznih bolesti u bolesnika s selektivnim nedostatkom IgA, zaraznih bolesti ENT organa i infekcija donjih dišni put... To je zbog činjenice da smanjenje sekretornog IgA, koji je dio lokalnog imuniteta, dovodi do lagane infekcije i razmnožavanja mikroorganizama na sluznicama, koje su najosjetljivije u dodiru s zarazne bolestiprenose se kapljicama u zraku.
U spektru nezaraznih bolesti pronađena je očita korelacija s autoimunim bolestima, koje su najvažnije manifestacije selektivnog nedostatka IgA, posebno s idiopatskom trombocitopeničnom purpurom (1,5-2 na 100 tisuća).
Od autoimune bolesti u bolesnika s selektivnim nedostatkom IgA najčešći su juvenilni reumatoidni artritis (4 puta), kronična idiopatska trombocitopenična purpura (3 puta) i autoimuni hepatitis (3 puta). Uz to, prema svjetskoj literaturi, bolesnici s selektivnim nedostatkom IgA imaju povećanu učestalost autoimunih stanja među najbližom rodbinom. No, prema našem istraživanju, njihov broj nije premašio opće vrijednosti populacije.
Incidencija atopijskih bolesti među bolesnicima s selektivnim nedostatkom IgA bila je značajno veća nego u populaciji (tablica 4). Samo je učestalost alergijskog rinitisa usporediva s općom populacijom. Takva se zapažanja odražavaju u nizu prethodnih studija. Ne može se reći da su alergijske bolesti u većine bolesnika s nedostatkom IgA teže nego u ljudi bez ovog imunološkog defekta. Međutim, velika prevalencija atopije postavlja pitanje provođenja imunološkog pregleda radi utvrđivanja oblika selektivnog nedostatka IgA, koji se još nisu klinički očitovali. Iako ovo možda neće imati odlučujuću ulogu u smislu pristupa terapiji za trenutno atopijsko stanje, pomoći će pravodobnoj dijagnozi i smanjenju mogućih rizika za ljude kod kojih će se otkriti selektivni nedostatak IgA.
U analizi ponovljenih imunograma tijekom dinamičkog promatranja u djece s selektivnim nedostatkom IgA, zbog trajnih promjena laboratorijskih parametara, dva su velike skupine bolesnika. Skupina A pokazala je odsutnost IgA bez ikakvih drugih promjena. U skupini bez IgA kombinirano je s trajnim porastom razine IgG. Provedena je usporedna analiza ovih skupina bolesnika.

Dob pojave kliničkih manifestacija u tim skupinama nije se značajno razlikovala.
Utvrđeno je da u bolesnika s selektivnim nedostatkom IgA, porast razine IgG korelira s ponavljajućim infekcijama kože i mekih tkiva. Ovo pitanje zahtijeva daljnje proučavanje.
Usporedbom ovih skupina bolesnika nisu utvrđene značajne razlike u spektru alergijske patologije.
Tijekom rada imunološki status procijenjen je u 20 obitelji bolesnika s selektivnim nedostatkom IgA. Utvrđena su 4 obiteljska slučaja. Uz to je prikupljena detaljna obiteljska povijest. Među odraslima rođaka s opterećenom zaraznom poviješću, koji su mogli provesti pregled, bilo je nekih kršenja humoralnog imuniteta. Sukladno tome, kada se otkriju manji humoralni nedostaci (posebno selektivni nedostatak IgA), ispitivanje srodnika, osobito u prisutnosti opterećene zarazne povijesti, obvezno je.
S obzirom na činjenicu da je selektivni nedostatak IgA među djecom iz dispanzerske promatračke skupine "često bolesna djeca" mnogo češći nego u općoj pedijatrijskoj populaciji, pedijatri koji se bave ovom bolešću moraju biti oprezni. Nije uvijek lako posumnjati na to, jer su kliničke manifestacije vrlo varijabilne: od asimptomatskih oblika do rekurentnih bakterijskih infekcija s potrebom za čestim antibakterijska terapija... Preporuča se proširiti znanje pedijatara i uskih specijalista u ambulantnoj i stacionarnoj njezi o manjim oštećenjima humoralnog imuniteta.
Budući da je među bolesnicima s selektivnim nedostatkom IgA učestalost alergijske patologije (bronhijalna astma, atopijski dermatitis, alergija na hranu) značajno veća, učestalost autoimunih bolesti i hematoloških bolesti, kao i učestalost kronična bolest (ORL organi, genitourinarni sustav, gastrointestinalnog trakta) nego u populaciji, njegova identifikacija je obavezna kako bi se osiguralo cjelovito i pravovremeno medicinska pomoć bolesnika.
Preporučuje se da se djeca s anamnezom zaraznih bolesti, pacijenti s hematološkim i autoimunim bolestima upućuju na konzultacije s imunologom / imunološkim pregledom, a treba provesti i probirni pregled ukupne razine IgA za pacijente s alergijskim bolestima.
Tijekom studije utvrđeno je da je većina djece s selektivnim nedostatkom IgA pokazala korelaciju između prisutnosti autoimuna patologija i trajni porast IgG u ponovljenim imunogramima. Za ostale bolesti takva korelacija nije pronađena. Takve promjene pokazatelja faktor su rizika za razvoj autoimune patologije u djeteta i zahtijevaju posebnu pozornost.
Unatoč činjenici da nije postojala korelacija između prisutnosti obiteljske anamneze selektivnog nedostatka IgA i ozbiljnosti kliničkih manifestacija u bolesnika, za te je bolesnike pregled najbližeg roda, posebno u prisutnosti opterećene zarazne anamneze, obvezan.
Književnost
- Hammarstrom L., Lonnqvist B., Ringden O., Smith C. I., Wiebe T. Prijenos nedostatka IgA na pacijenta s aplastičnom anemijom cijepljenom koštanom srži // Lancet. 1985; 1 (8432): 778-781.
- Latiff A. H., Kerr M. A.Klinički značaj nedostatka imunoglobulina A // Anali kliničke biokemije. 2007; 44 (Pt 2): 131-139.
- Al-Attas R. A., Rahi A. H.Primarni nedostatak antitijela u Arapa: prvo izvješće iz istočne Saudijske Arabije // Journal of Clinical Immunology. 1998; 18 (5): 368-371.
- Carneiro-Sampaio M. M., Carbonare S. B., Rozentraub R. B., de Araujo M. N., Riberiro M. A., Porto M. H.Učestalost selektivnog nedostatka IgA među brazilskim darivateljima krvi i zdravim trudnicama // Alergološka imunopatologija (Madr). 1989; 17 (4): 213-216.
- Ezeoke A. C. Selektivni nedostatak IgA (SIgAD) u istočnoj Nigeriji // African Journal of Medicine and Medical Sciences. 1988; 17 (1): 17-21.
- Feng L. Epidemiološka studija selektivnog nedostatka IgA među 6 nacionalnosti u Kini // Zhonghua Yi Xue Za Zhi. 1992; 72 (2): 88-90, 128.
- Pereira L. F., Sapina A. M., Arroyo J., Vinuelas J., Bardaji R. M., Prieto L. Prevalencija selektivnog nedostatka IgA u Španjolskoj: više nego što smo mislili // Krv. 1997; 90 (2): 893.
- Wiebe V., Helal A., Lefranc M. P., Lefranc G. Molekularna analiza multigene delegacije T17 imunoglobulina CH (del A1-GP-G2-G4-E) // Human Genetics. 1994; 93 (5): 5.
L.A. Fedorova *,
E. S. Puškova *
I. A. Korsunsky **, 1, Kandidat medicinskih znanosti
A.P. Prodeus *,doktor medicinskih znanosti, profesor
Imunodeficijencija - smanjenje kvantitativnih pokazatelja i / ili funkcionalne aktivnosti glavnih komponenata imunološki sustav, što dovodi do kršenja obrane tijela od patogenih mikroorganizama i očituje se povećanim zaraznim morbiditetom.
Kao što znate, glavna funkcija imunološkog sustava je prepoznavanje i uklanjanje stranih tvari antigene prirode koje prodiru u tijelo iz okoliš (mikroorganizmi) ili endogeno nastaju (tumorske stanice). Ova se funkcija provodi pomoću faktora urođeni imunitet (fagocitoza, antimikrobni peptidi, proteini komplementarnog sustava, NK-stanični sustav, itd.) i stečeni, odnosno adaptivni imunitet, provodi se pomoću staničnih i humoralnih imunoloških odgovora. Regulacija aktivnosti komponenata imunološke obrane tijela i njihova interakcija događa se uz pomoć citokina i međustaničnih kontakata.
U svakoj od navedenih komponenata imunološkog sustava, kao i u mehanizmima njihove regulacije, mogu se javiti poremećaji, što dovodi do razvoja imunodeficijencije, čija je glavna klinička manifestacija preosjetljivost na patogene zaraznih bolesti. Postoje 2 vrste imunodeficijencije: primarna i sekundarna.
Primarne imunodeficijencije(PID) - nasljedne bolestiuzrokovane nedostacima u genima koji kontroliraju imunološki odgovor. PID - bolesti, različite prirode i težine imunoloških defekata, kliničke manifestacije i molekularni poremećaji. Kliničku sliku PID karakteriziraju ponavljani i kronični teški zarazni procesi, uglavnom bronhopulmonalnog sustava.
i ENT organi, koža i sluznice; mogu se razviti gnojni limfadenitis, apscesi, osteomijelitis, meningitis i sepsa. U nekim oblicima postoje manifestacije alergija, autoimunih bolesti i razvoj nekih zloćudni tumori... Trebali biste obratiti pažnju na zaostajanje dobni pokazatelji tjelesni razvoj... Trenutno je opisano oko 80 PID-a i identificirani su geni odgovorni za razvoj većine ovih bolesti. Adekvatni laboratorijski testovi omogućuju razlikovanje patologije na razini limfocita i patologije na razini nelimfocitnih mehanizama uništavanja i eliminacije antigena.
Prevalencija PID-aovisi o obliku bolesti i u prosjeku se kreće od 1:10 000 do 1: 100 000 novorođenčadi. Selektivni nedostatak Primjerice, IgA je puno češći od 1: 500 do 1: 1500 u općoj populaciji. Rasprostranjenost različiti oblici PID varira u različite zemlje... Najčešći nedostaci u proizvodnji antitijela - 50-60% slučajeva, kombinirani PID - 10-30%, nedostaci fagocitoze - 10-20%, defekti komplementa - 1-6%. Većina PID-ova očituje se u ranom djetinjstvu, iako može doći do kasnijeg pojavljivanja nekih oblika PID-a, posebno uobičajenog varijabilnog imunološkog nedostatka (CVID).
Prema razvojnim mehanizmima, postoje 4 glavne skupine PID-a:
1. skupina - uglavnom humoralne ili B-stanice
PID;
Skupina 2 - kombinirani PID-ovi (kod svih imunodeficijencija T-stanica dolazi do disfunkcije B-stanica);
3. skupina - PID uzrokovani nedostacima u fagocitozi;
4. skupina - PID, uzrokovane nedostacima u sustavu komplementa.
Principi za dijagnozu primarnih imunodeficijencija
Rano dijagnosticiranje i pravovremeno započinjanje liječenja određuju prognozu bolesti. Postavljanje dijagnoze na razini okružnih pedijatara predstavlja određene poteškoće, što je često posljedica nedostatka mogućnosti pravovremenog savjetovanja pacijenta od strane imunologa i provođenja posebnog laboratorijskog imunološkog pregleda (tablica 11-1). Iako znanje o značajkama kliničke slike PID-a i promjenama
u općim kliničkim laboratorijskim testovima moguće je posumnjati na PID i uputiti pacijenta na specijaliste. Europsko društvo za imunodeficijencije razvilo je i stvorilo protokole za ranu dijagnozu PID-a elektronička baza podaci iz europskog PID registra. Dijagnostički algoritam PID prikazan je na sl. 11-1.
Tablica 11-1.Faze imunološkog pregleda u slučaju sumnje na imunodeficijenciju
Lik: 11-1.Algoritam za dijagnozu primarne imunodeficijencije
Opće značajke kliničke slike primarne imunodeficijencije
Vodeći u klinička slika PID je tzv infektivni sindrom - povećana osjetljivost na patogene zaraznih bolesti općenito, neuobičajeno teške ponavljajuće (ponavljajuće) ih klinički tijek, prisutnost atipičnih patogena (često oportunističkih) u etiologiji bolesti. Vrsta patogena određena je prirodom imunološkog defekta. U slučaju nedostataka u stvaranju antitijela, moguće je utvrditi otpornost na antibakterijski lijekovi flora - stafilokoki, streptokoki, Haemophilus influenzae. U imunološkom nedostatku T-stanica, osim bakterija, otkrivaju se i virusi (na primjer, obitelj herpesvirusa), gljivice (Candida spp., Aspergillusi drugi), te s fagocitnim defektima - stafilokokima, gram-negativnim bakterijama, gljivicama itd.
Laboratorijska istraživanja
Ako klinički dokazi ukazuju na PID, treba provesti sljedeće studije:
Određivanje detaljne krvne slike (posebno su važni kvantitativni i postotni pokazatelji limfocita);
Određivanje razina IgG, IgA i IgM u serumu;
Brojanje subpopulacija limfocita T i B;
◊ analiza funkcionalnog stanja fagocita (najjednostavnija i najinformativnija analiza je test za obnavljanje tetrazolium plavog);
◊ analiza sadržaja glavnih komponenata komplementa (započnite s C3 i C4);
◊ testiranje na HIV infekciju (ako postoje mogući faktori rizika);
Molekularno-genetske studije kada je naznačeno.
Principi liječenja primarnih imunodeficijencija
Glavni cilj PID terapije je liječenje i prevencija komplikacija bolesti. Ovaj je pristup rezultat činjenice da su nedostaci imunološkog sustava u PID-u na genetskoj razini. Trenutno se intenzivno istražuju geni
terapija imunodeficijencija, što može dovesti do pojave radikalnijih metoda njihovog liječenja.
Ovisno o obliku PID-a, liječenje se sastoji od supstitucijske terapije, liječenja i prevencije zaraznih, autoimunih manifestacija bolesti, liječenja malignih novotvorina i primjene posebne metode, uključujući transplantaciju krvotvornih matičnih stanica (ovisno o vrsti PID-a).
MANJKE IMUNOGLOBULINA
Prolazna hipogamaglobulinemija u djece
Privremena hipogamaglobulinemija u djece povezana je s fiziološkim obilježjem faznog stvaranja imunoglobulinskog sustava. Sazrijevanje stvaranja IgM i IgA antitijela najviše se "odgađa". U zdrave djece sadržaj majčinih IgG postupno se smanjuje i nakon šest mjeseci povećava se proizvodnja vlastitih IgG antitijela. Međutim, kod neke djece porast razine imunoglobulina je odgođen. Takva djeca mogu patiti od ponavljajućih bakterijskih zaraznih bolesti. U tim se slučajevima ne smije pribjegavati infuzijama donornog imunoglobulina (intravenski imunoglobulin).
Selektivni nedostatak imunoglobulina A
Selektivni nedostatak imunoglobulina A (SD IgA - Selektivni nedostatak IgA)razvija se kao rezultat defekta gena tnfrsf13b
ili p). Nedostatak IgA u prisutnosti imunoglobulina drugih klasa najčešća je imunodeficijencija otkrivena u općoj populaciji s učestalošću 1: 500-1500 ljudi (još češće u bolesnika s alergijama). Razlikovati selektivni nedostatak IgA, t.j. koji se sastoji u nedostatku jedne od potklasa (30% slučajeva) i potpune (70% slučajeva). Nedostatak u podrazredu IgA2 dovodi do izraženije kliničke slike od nedostatka u podrazredu IgA1. Moguće su i kombinacije nedostatka IgA s drugim poremećajima: s nedostatkom u biosintezi IgG i s abnormalnostima T-limfocita. Velika većina ljudi s selektivnim
nedostatak IgA je praktički zdrav. Za djecu mlađu od 2 godine nedostatak IgA je fiziološko stanje.
Smanjenje koncentracije IgA u serumu na<5 мг/дл у детей старше 4 лет; IgG и IgM в норме, количество и соотношение субпопуляций лимфоцитов и их функциональная активность могут быть в норме.
Klinička slika.S nedostatkom IgA mogu se razviti 3 skupine patoloških sindroma: infektivni, autoimuni i alergijski. Pacijenti s nedostatkom IgA skloni su ponavljajućim infekcijama gornjih dišnih putova i probavnog sustava. Najčešće i najteže su razne autoimune bolesti (reumatoidni artritis, ankilozirajući spondilitis, Sjogrenov sindrom, vaskulitis s oštećenjem cerebralnih žila, autoimuni tiroiditis, SLE, glomerulonefritis, hemolitička anemija, dijabetes melitus tipa I, vitiligo itd.) Incidencija celijakije za 10 puta prelazi onu u djece s normalnim IgA. Najčešće otkrivene alergijske manifestacije: netolerancija na proteine \u200b\u200bkravljeg mlijeka, atopijski dermatitis (AD), bronhijalna astma.
Liječenje.Asimptomatski slučajevi ne zahtijevaju nikakav poseban tretman; u prisutnosti kliničkih manifestacija zaraznih, autoimunih i alergijskih bolesti, liječenje se provodi u skladu sa standardima.
Zamjenska terapija donornim imunoglobulinima nije indicirana ni kod selektivnog ni kod potpunog nedostatka IgA, jer postoji velika vjerojatnost stvaranja antiizotipskih protutijela na IgA u primatelja i razvoja transfuzijskih komplikacija koje oni uzrokuju.
Agamaglobulinemija s nedostatkom B stanica
X-vezana agamaglobulinemija (Brutonova bolest)čini 90% svih slučajeva agamaglobulinemije. Dječaci, sinovi (אּ, ρ) nositelja neispravnog gena su bolesni btk (Xq21,3-q22),kodirajući B-limfocite specifičnu protein tirozin kinazu Btk (Brutonova tirozin kinaza- Brutonova tirozin kinaza). Kao rezultat defekta, poremećeni su unutarstanični signalni putovi, rekombinirani su teški lanci imunoglobulina,
pretvaranje pre-B-stanica u B-limfocite. U 10% bolesnika agamaglobulinemija s nedostatkom B-stanica nasljeđuje se na autosomno recesivni način. Trenutno je opisano 6 genetskih defekata, uključujući receptor za B-stanice, citoplazmatski protein B-stanični adapter (BLNK) i gen Leucinom bogato ponavljanje 8 (LRRC8).
Podaci laboratorijskih istraživanja.Nema perifernih B limfocita. Koštana srž sadrži pre-B stanice s μ-lancem u citoplazmi. Broj T-limfocita i testovi funkcije T-limfocita mogu biti normalni. IgM i IgA u krvi se ne mogu otkriti; IgG može biti prisutan, ali u malim količinama (0,4-1,0 g / l). Ne postoje antitijela na antigene krvnih grupa i antigene cjepiva (tetanus, toksini difterije itd.). Može se razviti neutropenija. Histološki pregled limfoidnog tkiva: u limfoidnim folikulima nema zametnih (embrionalnih) centara i plazma stanica.
Klinička slika.Ako nije poznata obiteljska povijest, dijagnoza postaje očita u prosjeku za 3,5 godine. Bolest je karakterizirana hipoplazijom limfoidnog tkiva, teškim gnojnim infekcijama, zaraznim bolestima gornjeg (sinusitis, otitis media) i donjeg (bronhitis, upala pluća) respiratornog trakta; mogući gastroenteritis, piodermija, septički artritis (bakterijski ili klamidijski), septikemija, meningitis, encefalitis, osteomielitis. Uzročnici respiratornih bolesti su najčešće Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus,proljev crijevne bakterije ili lamblije Giardia lamblia.Također, bolesnici s agamaglobulinemijom skloni su zaraznim bolestima uzrokovanim mikoplazmama i ureaplazmama, koje su uzrok razvoja kronične upale pluća, gnojnog artritisa, cistitisa i apscesa potkožnog tkiva. Tipični virusi su neurotropni virusi ECHO-19 i coxsackie, koji uzrokuju teški akutni i kronični encefalitis i encefalomielitis. Manifestacije enterovirusnih infekcija mogu biti sindrom sličan dermatomiozitisu, ataksija, glavobolje i poremećaji u ponašanju. U bolesne djece, kada se imunizira živim cjepivom protiv dječje paralize, u pravilu se otkriva produljeno prolijevanje virusa poliomijelitisa kroz sluznicu te s obnovljenom i sve većom virulencijom (tj.
postoji stvarna opasnost od zaraze zdrave djece poliomijelitisom kao rezultat kontakta s cijepljenim imunodeficijentnim djetetom). Autoimune poremećaje u agamaglobulinemiji mogu predstavljati reumatoidni artritis, sindrom sličan sklerodermu, skleredem, ulcerozni kolitis, dijabetes melitus tipa I (zbog prevladavanja imunološkog odgovora Th1).
Sistematski pregled.Obratite pažnju na zaostajanje u tjelesnom razvoju, na oblik prstiju (prsti u obliku bataka), promjene oblika prsnog koša, karakteristične za bolesti donjih dišnih putova, hipoplaziju limfnih čvorova i tonzila.
Liječenje.
Supstitucijska terapija: intravenski pripravci imunoglobulina primjenjuju se svaka 3-4 tjedna tijekom života. Doze imunoglobulina odabiru se tako da stvore koncentraciju u serumu pacijenta koja preklapa donju granicu dobne norme.
Razgovarajte o mogućnosti genske terapije - gen Btkklonirano, ali njegova prekomjerna ekspresija povezana je sa zloćudnom transformacijom krvotvornog tkiva.
U slučaju trajne neutropenije, koriste se faktori rasta. Kada se pojave znakovi autoimune patologije, moguće je propisati monoklonska antitijela (infliksimab, itd.).
Uobičajeni varijabilni imunološki nedostatak
Općenito varijabilni imunološki nedostatak (CVID) skupina je sindroma koju karakterizira nedostatak u sintezi antitijela i staničnom imunitetu. Pouzdan dijagnostički kriterij za CVID je značajno smanjenje sadržaja imunoglobulina dva ili tri glavna izotipa u oba spola u kombinaciji s jednim od sljedećih znakova:
Početak bolesti u dobi starijoj od 2 godine;
Nedostatak izohemaglutinina i / ili nizak odgovor na cijepljenje;
Isključenje drugih uzroka agamaglobulinemije.
U nekih su bolesnika uzrok razvoja CVID mutacije gena koji kodiraju molekule uključene u procese sazrijevanja i preživljavanja B stanica: BAFF-R (Receptor za aktiviranje faktora B-stanica),Blimp-1 (B-limfociti izazvani sazrijevanjem protein-1)i ICOS (Inducibilni kostimulator).Dolazi do kršenja sposobnosti B-limfocita da se diferenciraju u plazma stanice, razvijaju se nedostaci u stvaranju antitijela, moguća je disfunkcija T-limfocita i uočava se povećana sklonost zaraznim bolestima. Sindrom se može manifestirati u ranom djetinjstvu, adolescenciji ili mladim odraslima.
Podaci laboratorijskih istraživanja.Razine IgG i IgA (u oko 50% bolesnika) i IgM (do neotkrivenih količina) značajno su smanjene. Broj B-limfocita u krvi je normalan ili smanjen. Broj T-limfocita u većine je bolesnika normalan. U ozbiljnih bolesnika može se razviti limfopenija (manje od 1500x10 3 stanice u 1 litri krvi). Broj NK stanica je smanjen. Proizvodnja specifičnih antitijela kao odgovor na imunizaciju smanjena je ili odsutna. Proliferacija limfocita i stvaranje IL-2 pod utjecajem mitogena i antigena znatno su oštećeni.
Klinička slika.Otkrivaju se ponavljajuće bakterijske zarazne bolesti s lokalizacijom uglavnom u respiratornom traktu i paranazalnim sinusima. U vrijeme dijagnoze infekcije respiratornog trakta mogu preći u bronhiektazije i difuzne lezije plućnog tkiva. Moguća je zarazna lezija probavnog sustava, koja se očituje proljevom, steatorejom i malapsorpcijom (i, shodno tome, gubitkom težine). Infekcije uzrokovane Giardia lamblia, Pneumocystis cariniiili obiteljske viruse Herpetoviridae.Pacijenti s CVID-om skloni su razvoju gnojnog artritisa uzrokovanog mikoplazmama i ureaplazmama. Manifestacije enterovirusnih infekcija mogu biti encefalomielitis, poliomijelitis i sindromi slični dermatomiozitisu, lezije kože i sluznice. Autoimunebolesti su teške i mogu odrediti prognozu CVID-a. Ponekad su prve kliničke manifestacije CVID artritis, ulcerozni kolitis i Crohnova bolest, sklerozirajući holangitis, malapsorpcija, SLE, nefritis, miozitis, autoimuna bolest pluća u obliku limfoidnog intersticijskog pneumonitisa, neutropenija,
trombocitopenična purpura, hemolitička anemija, perniciozna anemija, alopecija totalis, vaskulitis mrežnice, fotosenzibilnost. U bolesnika s CVID-om učestalost (u 15% slučajeva) granuloma sličnih sarkoidozi i nemaligne limfoproliferacije je značajno povećana. Liječenje.
Antibakterijska kemoterapija.
Supstitucijska terapija: intravenski pripravci imunoglobulina daju se svaka 3-4 tjedna tijekom života.
U slučaju autoimunih komplikacija - imunosupresivna terapija (glukokortikoidi, azatioprin, ciklosporin A) i eventualno imenovanje monoklonskih antitijela (infliksimab, itd.).
Hiper-IgM sindromi
Hiper-IgM sindromi prilično su rijetke bolesti koje karakterizira izraženo smanjenje ili potpuno odsustvo IgG, IgA i normalna ili povećana koncentracija IgM u serumu. To je zbog nemogućnosti B-limfocita da prebace klase imunoglobulina i hipermutageneze varijabilnih domena. Do danas je identificirano 6 genetskih nedostataka koji dovode do razvoja hiper-IgM sindroma.
. Tip 1 (HIGM 1).X-povezani nedostatak CD40 liganda (70% slučajeva hiper-IgM sindroma), što dovodi do nesposobnosti T stanica da učinkovito komuniciraju s B limfocitima.
. Tip 2 (HIGM 2).Autosomno recesivno, povezano s aktivacijom citidin deaminaze izazvanom AID defektom (gen Aicda, 12p13)- enzim koji sudjeluje u izmjeni klasa imunoglobulina i hipermutagenezi.
. Tip 3 (HIGM 3).Autosomno recesivno, povezano s genskom mutacijom molekule CD40. U ovom slučaju, B stanice same po sebi nisu u mogućnosti učinkovito komunicirati s T limfocitima. Fenotipske manifestacije slične su onima tipa 1.
. Tip 4 (HIGM 4).Autosomno recesivno; u nekim se slučajevima javljaju mutacije de novo.Povezan s defektom u UNG - uracil-DNA glikozilaza - enzim koji je također uključen u
u zamjenjivanju klasa imunoglobulina, ali nakon djelovanja AID-a. U ovom slučaju, hipermutageneza nije pogođena i sindrom teče s manje ozbiljnosti.
. Tip 5 (HIGM 5).Oštećena samo prebacivanjem klase, hipermutageneza nije pogođena. Uzročna mutacija još nije utvrđena, ali očito postoji nedostatak u enzimu koji djeluje nakon toga
POMOĆ.
. Tip 6 (HIGM-ED).X-povezano, povezano s dishidrotskom ektodermalnom displazijom, uzrokovano je nedostatkom NEMO (NF-kB modulatora), što rezultira oslabljenom CD40 signalizacijom.
X-povezani hiper-IgM sindromidentificirani češće od ostalih. Razvija se s nedostatkom u genu koji kodira CD40L (CD154, gen je smješten na Xq26-q27.2)je ligand za CD40. Nedovoljna ekspresija CD40L od strane T-limfocita dovodi do nemogućnosti prebacivanja klase imunoglobulina u B-limfocitima s IgM na druge izotipe, kao i do poremećenog stvaranja memorijskih B-stanica, repertoara T-stanica i odgovora Th1-stanica usmjerenih protiv unutarstaničnih mikroorganizama. Dječaci su bolesni
Podaci laboratorijskih istraživanja.IgG, IgA, IgE nije moguće odrediti ili se otkrivaju u vrlo malim količinama. Razine IgM su normalne (u 50% slučajeva) ili povišene, često značajno. Broj T i B stanica je normalan; smanjeni proliferativni odgovor T stanica, induciran antigenima. IgM su poliklonski, ponekad i monoklonski. Otkriti autoantitijela izotipa IgM (anti-eritrociti, antitrombociti, antitiroidni, antitijela na antigene glatkog mišićnog tkiva). U limfoidnom tkivu nema zametnih centara, ali postoje plazma stanice.
Klinička slika.Prve manifestacije javljaju se u djetinjstvu i ranom djetinjstvu. Ponovljeno infekcijerazličita lokalizacija (prvenstveno dišni put), uključujući oportunističku (uzrokovanu Pneumocystis carinii).Također su karakteristične lezije virusima (citomegalovirus i adenovirusi), Criptococcus neoformans,mikoplazme i mikobakterije. Kriptosporidijalna infekcija može uzrokovati akutni i kronični proljev (razvija se u 50% bolesnika) i sklerozirajući kolangitis. Anemija, neutropenija, ulceracija usne sluznice, gingivitis, ulcerativni
lezije jednjaka, razni dijelovi crijeva, ulcerozni kolitis. Otkrivaju sklonost ka autoimuni poremećaji(seronegativni artritis, glomerulonefritis, itd.) i maligne novotvorine (uglavnom limfoidno tkivo, jetra i žučni trakt). Mogu se razviti limfadenopatija, hepato- i splenomegalija. Liječenje
Redovita nadomjesna terapija intravenskim imunoglobulinom.
Antibakterijska kemoterapija. Za prevenciju i liječenje pneumocystis upale pluća koriste se ko-trimoksazol [sulfametoksazol + trimetoprim] i pentamidin.
Da biste spriječili oštećenje jetre i žučnih puteva, trebali biste koristiti samo prokuhanu ili filtriranu vodu, provoditi redovite preglede (ultrazvuk, biopsija jetre ako je naznačeno).
U liječenju neutropenije i oralne ulceracije koriste se glukokortikoidi i pripravci koji stimuliraju koloniju granulocita.
S razvojem autoimunih komplikacija propisana je imunosupresivna terapija (glukokortikoidi, azatioprin, ciklosporin A), kao i lijekovi na bazi monoklonskih antitijela.
Optimalna metoda liječenja je transplantacija koštane srži od davatelja koji se podudaraju s HLA-om (stopa preživljavanja 68%, najbolje učiniti prije 8. godine).
KOMBINIRANE IMUNODEFECIJENCIJE SA PREVENTIVNIM DEFEKTOM T-LIMFOCITA
Teški kombinirani imunološki nedostatak
SCID (SCID - Teški kombinirani imunološki nedostatak)- skupina sindroma koju karakterizira smanjenje razine T-limfocita ili njihovo potpuno odsustvo i kršenje adaptivnog imuniteta. ... Retikularna disgeneza, karakterizirana oštećenim sazrijevanjem limfoidnih i mijeloidnih prekursora u ranim fazama: neutropenija i T - B - NK -.
. X-povezani SCID, koji se razvija kao rezultat mutacije gena IL-2RG[(CD132, ukupno na- lanac receptora za IL-2, IL-4, IL-7, IL-9, IL-15 i IL-21), Xq13,1-q21,1,אּ ], što dovodi do blokade receptora i nemogućnosti ciljnih stanica da odgovore na djelovanje odgovarajućih interleukina (više od 50% svih slučajeva SCID-a); T - B + NK -.
. Janus3 nedostatak tirozin kinaze [gen JAK3 (19p13,1),ρ ]; s defektima gena, prijenos aktivacijskog signala od općeg na-lanci IL-2, IL-4, IL-7, IL-9, IL-15, IL-21 do stanične jezgre, što dovodi do poremećene diferencijacije T- i NK-stanica; T - B + NK -.
. Nedostatak proteinske tirozin fosfataze (CD45, gen PTPRC, 1q31-q32);s genskim defektom, dolazi do povećanja inhibitorne aktivnosti Csk kinaze na protein tirozin kinazu Src s oštećenom fosforilacijom ITAM domena TCR i BCR; T - B + NK +.
. Potpuni nedostatak enzima RAG1 i RAG2, koji aktiviraju rekombinaciju V (D) J-segmenata imunoglobulina i TCR [gena RAG1i RAG2 (11p13),ρ ]; T - B - NK +.
. Omennov sindrom (nepotpuni nedostatak RAG1 i
RAG2) [geni RAG1i / ili RAG2 (11p13-p12),r].Zahvaljujući
niska rezidualna aktivnost ovih enzima i dalje razvija brojne klonove T-limfocita, specifičnih za antigene epitelnih tkiva kože i probavnog trakta, gdje se množe i proizvode velike količine IL-4 i IL-5, što uzrokuje hipereozinofiliju i stvaranje IgE od zaostalih B-limfocita (u nedostatku drugih klasa imunoglobulina). Karakterizirana eritrodermijom i pahidermijom s alopecijom u vlasištu i obrvama, iscrpljujućim proljevom, zaraznim sindromom opasnim po život; hepatosplenomegalija i hiperplazija limfnih čvorova.
. SCID s povećanom osjetljivošću na ionizirajuće zračenje. Defekt nuklearnog proteina Artemis [gen DCLRE1C, (10p),r],uključen u kompleks enzima neophodnih za popravak DNA (sudjeluje u povezivanju dvolančanih prekida), s genskom mutacijom, dolazi do kršenja V (D) J-rekombinacije; T - B - NK +.
. Nedostatak IL-2 [gen IL-2, 4q26-q27].
Mutacije u genu lanca receptora za IL-2 (CD25) (10p15-p14);T - B + NK +.
Mutacije u genu lanca receptora za IL-7 (CD127) (5p13);T - B + NK +.
Nedostatak TAP-a (Transporter za prezentaciju antigena),potreban za transport antigenih peptida u endoplazmatski retikulum lanca gena receptora IL-7 (CD127) (5p13);T - B + NK +.
Mutacije u genima CD3 lanca (CD3γ, CDδ i CDε), što dovodi do smanjenja broja zrelih T-limfocita, oštećenja njihove diferencijacije; T - B + NK +.
Nedostatak proteinske tirozin kinaze ZAP-70 [gen ZAP-70 (2q12),r]. Uz gensku mutaciju trpi fosforilacija ITAM domena ζ-lanca TCR-a i receptora NK stanica koji sadrže ITAM, a razvija se i selektivni nedostatak CD8 + T stanica (sadržaj CD4 + T limfocita je normalan, ali funkcionalni poremećaji izraženi su u obliku odsutnosti IL- 2 i širenje).
Nedostatak adenozin deaminaze [gen ada (20q12-q13.11 , p)], što dovodi do nakupljanja metabolita u stanicama (deoksiadenozin trifosfat i S-adenosil homocistein) koji inhibiraju proliferaciju T- i B-limfocita (opisane su inačice s kasnim početkom bolesti); T - B - NK -.
Nedostatak purinske nukleozidne fosforilaze [gen pnp (14q11,2),p], što dovodi do nakupljanja deoksiguanozin trifosfata u stanicama, što inhibira proliferaciju T-limfocita (popratni sindromi - uricemia i uricuria); T - B + NK -.
Podaci laboratorijskih istraživanja.Otkrivanje varijabilne, ponekad duboke limfopenije; limfociti se ne mogu razmnožavati kao odgovor na specifični antigen; često se izražava smanjenje razine imunoglobulina u krvnom serumu. Na rendgenu prsnog koša nema sjene timusa.
Klinička slika.Klinička dijagnoza obično postaje jasna u prvih 6 mjeseci života, kada majčina IgG antitijela nestanu. U kliničkoj slici dođite do izražaja teški zarazni sindrom,hipoplazija limfoidnog tkiva i zastoj u razvoju. Infektivni sindrom karakterizira oralna kandidijaza, kronični proljev, upala pluća, vrućica,
sepsa bakterijske etiologije, virusne infekcije. Uzročnici infekcija pripadaju različitim taksonomskim skupinama: bakterija, virusa, gljivica, oportunističkih mikroorganizama (Pneumocystis carinii).Upala pluća često je uzrokovana P. carinii,proljev - rotavirusi, Campylobacter, Giardia lamblia.Virusni hepatitis se često manifestira. Karakterističan je razvoj regionalnog ili generaliziranog BCGitisa nakon cijepljenja.
Liječenjepredviđa imenovanje suportivne terapije, uključujući parenteralnu prehranu, uvođenje intravenskog imunoglobulina, imenovanje antibiotika, antifungalnih i antivirusnih lijekova. Jedna od glavnih metoda liječenja za postizanje oporavka je transplantacija koštane srži, bez koje djeca s SCID-om obično umiru u prvoj godini života. Opisani su izolirani slučajevi kada je dijete u posebno saniranim uvjetima živjelo do 2-3 godine. Važno je prepoznati SCID u novorođenčadi što je ranije moguće, jer je, na primjer, imunizacija živim cjepivima za njih kobna. Neposredno nakon postavljanja dijagnoze, takvu djecu treba smjestiti u gnotobiološke uvjete (sterilna kutija). U slučaju dodavanja zaraznih bolesti, provodi se intenzivna antibakterijska, antivirusna i antifungalna terapija, zamjenska terapija intravenskim imunoglobulinom. Za prevenciju pneumocistične upale pluća, propisuje se kotrimoksazol. U slučaju razvoja BCGitisa potrebno je provoditi dugotrajnu intenzivnu antituberkuloznu terapiju. Za transfuziju komponenata krvi smiju se koristiti samo ozračeni i filtrirani lijekovi. Postoji rizik od razvoja reakcije transplantata nasuprot domaćina zbog transplacentarnog prijenosa majčinih limfocita.
Sindrom golih limfocita
Ovo je naziv patologije kada tijelo ne eksprimira molekule MHC-I ili MHC-II. U nedostatku ekspresije molekula MHC-I, sadržaj CD8 + T-limfocita je smanjen i nema aktivnosti NK-stanica; u odsustvu MHC-II, razina CD4 + T-limfocita je smanjena. Okarakterizirano je nekoliko genetskih nedostataka. Međutim, ovi nedostaci nisu lokalizirani u MHC genima, već u nekoliko različitih čimbenika koji su odgovorni za njihovu regulaciju
izraz. Klinička slikasindrom "golih" limfocita i liječenjeslični su onima za druge SCID-ove.
DiGeorgeov sindrom
S DiGeorgeovim sindromom ili sindromom trećeg i četvrtog ždrijelnog džepa [brisanja u 22q11,uključujući gen TBX1 (22q11,2),otkrivaju hipoplaziju ili aplaziju timusa, hipoplaziju paratireoidne žlijezde, srčane mane, nedostatak T-limfocita, promjenjivi broj B-limfocita.
Podaci laboratorijskih istraživanja.Značajno smanjenje broja CD3 +, CD4 + i CD8 + T stanica i naglo smanjenje njihove proliferativne aktivnosti inducirane mitogenima i antigenima. Broj B i NK stanica je normalan. Koncentracija serumskih imunoglobulina u većini je slučajeva u granicama normale, moguće su razne varijante disgamaglobulinemije.
Klinička slika.Komponenta imunodeficijencije predstavljena je hipoplazijom ili aplazijom timusa i ponavljajućom, teškom zarazne bolesti.Također se otkriva hipoparatireoidizam (hipokalcemija i, kao posljedica toga, tetanija, primjetna 1-2. Dana nakon rođenja); defekti krvožilnog sustava (desni zavoj luka aorte, stenoza desne klijetke, defekti interventrikularne i interatrijske pregrade, tetrad Fallota, atrezija ili hipoplazija plućne arterije); udubljenje nepca; anomalije kostura lica (povećana udaljenost između uparenih organa, čeljusti smanjene veličine, posebno donji, nisko postavljeni uši, kratki nosni žlijeb). Izražene su abnormalnosti strukture grkljana, ždrijela, dušnika, unutarnjeg uha, jednjaka; oslabljen razvoj bubrega, središnjeg živčanog sustava i druge malformacije (polidaktilija, odsutnost noktiju, atrezija anusa, analne fistule). Karakteristično je kašnjenje u govoru i psihomotornom razvoju. Primjećuju sklonost ka autoimuni poremećaji(citopenija, autoimuni tiroiditis) i maligne novotvorine.
Liječenje.... Antibakterijska i antivirusna terapija. ... Supstitucijska terapija intravenskim pripravcima imunoglobulina. ... Kirurško liječenje radi ispravljanja malformacija. ... Za autoimune komplikacije - imunosupresivna terapija. ... U prisutnosti endokrinopatija, korekcija odgovarajućih poremećaja. ... Transplantacija koštane srži je neučinkovita
tivna. ... Transplantacija epitelnog tkiva timusa je opravdana. Korekcija funkcije paratireoidnih žlijezda.
X-vezani limfoproliferativni sindrom
X- povezani limfoproliferativni sindrom karakterizira oslabljeni imunološki odgovor na Epstein-Barrov virus [uzrokovan genskim defektima SH2D1A(SAP) u Xq25,אּ], što dovodi do nekontrolirane proliferacije B-limfocita transformiranih virusom Epstein-Barr i zaraze novih ciljnih stanica virusom.
Klinička slika.Opisana su 4 najčešća fenotipa: teška infektivna mononukleoza, maligni limfoproliferativni uvjeti (limfomi, leukemije, uglavnom B-stanice), anemija ili pancitopenija (uključujući one zbog hemofagocitnog sindroma izazvanog virusom), disgamaglobulinemija. Infekcija virusom Epstein-Barr pokretački je (okidački) mehanizam za stvaranje najtežih, brzo progresivnih i fatalnih bolesti: fulminantna infektivna mononukleoza (fatalna u 58% slučajeva), hemofagocitni sindrom (bez liječenja, u 100% slučajeva smrtonosno). U 10% slučajeva fenotip se očituje prije infekcije virusom Epstein-Barr (s disgamaglobulinemijom i u pravilu se razvijaju limfomi). Najčešće se otkrivaju razne vrste hipogamaglobulinemije. Imunodeficijencija dovodi do razvoja bakterijskih, gljivičnih i virusnih zarazne bolesti.Moguće je pretpostaviti bolest kod dječaka s karakterističnom obiteljskom anamnezom i sero- ili PCR pozitivnim testom Epstein-Barr virusa. Za dijagnozu se preporučuje uporaba kombinacije genetske analize SH2D1Ai procjena razine izraženosti SAP-a.
Liječenje
U svrhu prevencije preporuča se uporaba antivirusnih lijekova - aciklovira, valaciklovira (njihova rana primjena suzbija replikaciju Epstein-Barrovog virusa u orofarinksu) i intravenskog imunoglobulina (s visokim titrom antitijela na Epstein-Barrov virus).
Za hipogamaglobulinemiju, intravenski imunoglobulin koristi se mjesečno u kombinaciji s antibiotskom terapijom.
Za liječenje fulminantne zarazne mononukleoze propisane su visoke doze aciklovira i metilprednizolona, \u200b\u200bvisoka doza terapije intravenskim imunoglobulinom s visokim titrom antitijela na Epstein-Barrov virus i IFNa.
S razvojem hemofagocitnog sindroma, visoke doze deksametazona kombiniraju se s vepezidom ♠ (etopozidom).
U liječenju malignih bolesti koriste se standardni protokoli terapije.
Radikalni tretman je transplantacija koštane srži od davatelja koji se podudaraju s HLA-om.
Autoimuni limfoproliferativni sindrom
Autoimuni limfoproliferativni sindrom skupina je bolesti koju karakteriziraju benigna limfoproliferacija, hiperimunoglobulinemija, autoimuni poremećaji, povećani sadržaj CD3 + CD4 - CD8 - T-limfociti (dvostruko negativni) u perifernoj krvi i defekt apoptoze [genski defekt Fas(CD95) - TNFRSF6 (10q24,1),gen kaspaza-10,Fas ligand - FasL (1q23)].
Podaci laboratorijskih istraživanja.Sadržaj CD3 + CD4 - CD8 - T-limfocita u perifernoj krvi ili limfoidnim tkivima je više od 1%. Razine IgG, IgA i IgM mogu biti normalne, povišene ili čak smanjene. S godinama hipergamaglobulinemija zamjenjuje se niskom koncentracijom serumskih imunoglobulina, sve do agamaglobulinemije. Otkriti autoantitijela na eritrocite, trombocite, neutrofile, glatke mišiće, na faktor VIII; antinuklearna i antifosfolipidna autoantitijela, kao i reumatoidni faktor itd. Karakteristična je limfocitoza.
Klinička slika.Svi pacijenti imaju povećanu jetru, limfne čvorove (u prvih 5 godina života) i slezenu. Limfoproliferaciju ne prati groznica i noćno znojenje. Debi autoimune reakcijene mora se podudarati s limfoproliferacijom i javlja se kasnije. S godinama se ozbiljnost autoimunih reakcija povećava. Češće se razvijaju autoimune reakcije protiv krvnih stanica (hemolitička anemija, trombocitopenija, neutropenija), rjeđe su zahvaćeni drugi organi. Povećan je rizik od razvoja malignih novotvorina (T i B limfomi, Burkittov limfom, atipični limfom, limfogranulomatoza, itd.).
Liječenje.... Kemoterapijska sredstva (ciklofosfamid, azatioprin, metotreksat, klorambucil). ... Glukokortikoidi. ... Splenektomija s jakim hipersplenizmom i hemocitopenijom. ... U težim slučajevima moguća je transplantacija koštane srži.
Hiperimunoglobulinemijski sindrom E
Hyper-IgE sindrom karakterizira značajan porast razine serumskog IgE, ponovljeni apscesi kože i potkožnog tkiva stafilokokne etiologije, upala pluća s nastankom pneumokele, anomalije u strukturi kostura lica, AD. Molekularno genetska priroda hiper-IgE sindroma još nije utvrđena. U nekim je slučajevima otkrivena autosomno dominantna, u drugima - autosomno recesivno nasljeđivanje. Pretpostavlja se da defekti utječu na signalne molekule citokinskih receptora (u autosomno dominantnom obliku ovog sindroma, mutacije u Stat3)i, moguće, povezano s disfunkcijom subpopulacije Th17. Drugi gen odgovoran za stvaranje hiper-IgE sindroma lokaliziran je na kromosomu 4 (4q).
Podaci laboratorijskih istraživanja.Otkrivaju se razni imunološki poremećaji: porast razine IgE u serumu, kršenje kemotaksije neutrofila, kvar u stvaranju antitijela; smanjenje odgovora HRT-a na toksoid kandida, difterije i tetanusa; slabljenje proliferativne aktivnosti T stanica kao odgovor na Candidai tetanusni toksoid zadržavajući odgovor na mitogene. Eozinofilija u perifernoj krvi i tekućini kožnih apscesa. Broj T i B stanica je normalan.
Klinička slika.Umjereni ekcem u ranoj dobi. Karakteristične crte lica (široki most na nosu, široki donji nos, asimetrija kostura lica, izbočeno čelo, duboko postavljene oči, visoko nepce). Otkrivaju se anomalije u razvoju kostura, skolioza, povećana pokretljivost zglobova, sklonost prijelomima kostiju nakon manjih ozljeda i kršenje zamjene zuba. Postoje apscesi kože, potkožnog tkiva i limfnih čvorova. Upala pluća razvija se u starijoj dobi (najčešći patogeni S. aureusi H. u-
gripa),u 77% slučajeva nastaje pneumokela, infekcija zbog P. aeruginosai A. fumigatus.Upala pluća može se odvijati bez temperature. Kronična kandidijaza sluznice i noktiju razvija se u 83% slučajeva.
Liječenje.... Dugotrajna (u svrhu prevencije - doživotna) antibakterijska i antifungalna terapija. ... Za liječenje dermatitisa koriste se lokalna sredstva, u težim slučajevima, male doze ciklosporina A. Transplantacija koštane srži je neučinkovita.
SINDROMI KROMOSOMSKOG KVARA
Za sindrome s kromosomskom nestabilnošću: ataksiateleangiectasia[defekt u genu DNA topoizomeraze Bankomat (11q22),p] i nijmegenov sindrom[Defekt gena Nibrin NBS1(8q21)] - karakterizirana povećanom učestalošću malignih tumora, spontanom kromosomskom nestabilnošću i kromosomskim raspadima. Oba proteina sudjeluju u popravljanju dvostrukih lanaca DNA i u regulaciji staničnog ciklusa. Dvolančani prelomi DNA obično se javljaju tijekom V (D) J rekombinacije imunoglobulina i TCR gena, mijenjajući klase imunoglobulina, tijekom križanja i tijekom mejoze. Slični se procesi događaju tijekom sazrijevanja neurona u mozgu. Defekti popravljanja DNA u ataxia-telangiectasia i Nijmegenovom sindromu uzrokuju takve kliničke manifestacije poput poremećaja u sintezi imunoglobulina, funkcije genitalnih organa i živčanog sustava.
Ataxia-telangiectasia
Ovaj sindrom (učestalost 1: 300 tisuća novorođenčadi) s vrlo heterogenim fenotipom opisao je francuski liječnik D. Louis-Bar. Simptomi ataksije mogu se otkriti kod djeteta već u dobi od 2-4 mjeseca. Ataxia je uzrokovana progresivnom degeneracijom Purkinjeovih stanica u malom mozgu. Telangiektazije na koži nosa, ušnim ušima i konjunktivi pojavljuju se nešto kasnije, za 3-6 godina. Često se na koži pojave mrlje od kave s mlijekom. Karakteristična je hipoplazija timusa, limfnih čvorova, slezene, tonzila. Imunodeficijencija se očituje smanjenjem (često neravnotežom) u proizvodnji IgA, IgE, IgG2, IgG4. 80% bolesnika se razvija
postoji odgovarajuća infektivna klinička simptomatologija. Smanjen broj i funkcionalna aktivnost T stanica (uglavnom CD4 + T stanica). Ukupni broj T limfocita je u većine bolesnika normalan. Neobično visoka (200 puta veća nego u općoj populaciji) učestalost novotvorina (uglavnom limfoma i karcinoma), što često dovodi do smrti za 10-12 godina. Liječenjesimptomatski.
Nijmegenov sindrom
Nijmegenov sindrom (imenom grada u Nizozemskoj, gdje je bolest prvi put opisana) očituje se mikrocefalijom, specifičnim poremećajima kostura lica (koso čelo, izbočeni srednji dio lica, dugačak nos, hipoplazija donje čeljusti, mongoloidni rez na oku, epikantus, velike uši), usporen fizički razvoj , prisutnost mrlja "kava s mlijekom" na koži; klinodaktilija i sindaktilija, disgeneza jajnika itd. Većina djece pati od ponavljajućih i kroničnih bakterijskih zarazne bolestirespiratorni trakt, ORL organi i mokraćni sustav. U 50% slučajeva razvijaju se maligne novotvorine, uglavnom limfomi B-stanica. Otkrivaju razne oblike disgamaglobulinemije, smanjenje CD4 + T stanica.
Liječenje.... Simptomatska terapija za neurološke poremećaje. ... Zamjenska terapija intravenskim imunoglobulinom. ... Prema indikacijama koristi se antibakterijska, antivirusna, antifungalna terapija. ... Pri liječenju malignih novotvorina uzima se u obzir povećana osjetljivost na zračenje i kemoterapiju.
Wiskott-Aldrichov sindrom
Wiskott-Aldrichov sindrom [defekt gena WASP (Xp11.23p11.22),אּ; također ρ i Ʀ] Gen OSA(iz Wiskott-Aldrichov sindrom)izraženo u limfocitima, tkivu slezene i timocitima. Mutacije u ovom genu povezane su s abnormalnom ekspresijom u neutrofilima i T-limfocitima (CD4 i CD8) molekule CD43 (ligand za ICAM-1, ima antiadhezivnu funkciju).
Podaci laboratorijskih istraživanja.Trombocitopenija (manje od 10% norme) uzrokovana je povećanim uništavanjem stanica.
Trombociti su manji nego u zdravih ljudi. Razina serumskog IgM smanjuje se s normalnom razinom IgG i porastom IgA i IgE. Smanjen titar izohemaglutinina, oslabio je stvaranje antitijela na polisaharidne antigene pneumokoka, streptokoka, Escherichia coli, Salmonelle, kao i antivirusna antitijela. U ranoj dobi, u pravilu, broj limfocita je normalan; nakon 6 godina otkriva se limfopenija (manja od 1x10 9 / l), smanjenje CD3 + i CD4 + T stanica s normalnom razinom B i NK stanica. Moguća je eozinofilija i razvoj posthemoragične anemije. Proliferativni odgovor T stanica na mitogene i antigene se smanjuje, HRT je oslabljen. U slezeni se ne otkrivaju normalne strukture zametnih centara i zona T-stanica.
Klinička slika.Bolest karakterizira trijada simptoma: trombocitopenija, ekcem i ponavljajuće zarazne bolesti.Hemoragijski sindrom manifestira se rano, već u novorođenčetu (petehijalni osip, cefalohematomi, krvarenje iz pupkovine, crijevna krvarenja). Ekcem se očituje od najranije dobi u 80% bolesnika. Znakovi imunodeficijencije povećavaju se s godinama: bakterijske zarazne bolesti ENT organa, dišnog sustava, probavnih organa, kože; česta ili generalizirana herpes infekcija (Herpes simplexi Varicella zoster),citomegalovirus, kao i gljivične (kandidijaza sluznice), rjeđe oportunističke zarazne bolesti. Autoimune bolesti (hemolitička anemija, neutropenija, artritis, kožni vaskulitis, ulcerozni kolitis, cerebralni vaskulitis, glomerulonefritis, autoimuna trombocitopenija) dijagnosticiraju se u 70% bolesnika. U bolesnika starijih od 5 godina, učestalost maligne novotvorine(uglavnom tumori limfoidnog tkiva).
Liječenje.... Alogena transplantacija koštane srži ili matičnih stanica (stopa uspješnosti operacije doseže 90% kada se koristi transplantat histokompatibilnog davatelja i 50% kod haploidentične transplantacije). ... Zamjenska terapija intravenskim imunoglobulinom. ... Preventivna primjena antibakterijskih, protugljivičnih i antivirusnih lijekova. ... Da bi se smanjio hemoragični sindrom, vrši se splenektomija. ... Za autoimune komplikacije propisana je imunosupresivna terapija.
MANJKE FAGOCITOZE
Kronična granulomatozna bolest
Kroničnu granulomatoznu bolest karakteriziraju poremećena funkcionalna aktivnost fagocita (stvaranje aktivnih oblika radikala kisika, unutarstanično ubijanje i fragmentacija fagocitoziranih patogena), trajne bakterijske i gljivične zarazne bolesti te razvoj granulomatozne upale. Kronična granulomatozna bolest razvija se u osoba s različitim genetskim nedostacima [u 65% slučajeva - X-vezana varijanta bolesti: gen gp91-phox (Xp21.1),אּ; u 35% slučajeva - autosomno recesivno: gen f47-phox (7q11,23),ρ; gen p67-foks (1q25),ρ; gen p22-phox (16q24),p], što dovodi do poremećaja u sustavu NADP-oksidaze. Smrću kratkotrajnih (nekoliko sati) neutrofila, neuništene bakterije "utječu" u žarište upale. Makrofagi su dugovječne stanice i njihovi prethodnici (monociti) u povećanom broju migriraju u fokus (što dovodi do stvaranja granulomi),fagocitozni mikroorganizmi, ali ih nisu u stanju ubiti.
Podaci laboratorijskih istraživanja.Imunoglobulini u serumu i subpopulacije limfocita su normalne. Stvaranje peroksidnih radikala od strane neutrofila, procijenjeno u testovima (kemiluminescencija ovisna o luminolu ili redukcija tetrazolium plavog), naglo je smanjeno ili odsutno. U pozadini zaraznih bolesti karakteristični su leukocitoza, neutrofilija, povećana ESR, anemija i hipergamaglobulinemija.
Klinička slika.Bolest se u većini slučajeva očituje u prvoj godini života. infektivni sindrom(infekcije unutar- i izvanstaničnim patogenima) i stvaranje granuloma. Najtipičnije: oštećenje pluća (ponovljena upala pluća, oštećenje hilarnih limfnih čvorova, apscesi pluća, gnojni pleuritis), probavnog trakta, apscesi kože i limfadenitis. Najčešći patogeni su mikroorganizmi pozitivni na katalazu: S. aureus, Aspergillus spp.,crijevne gram negativne bakterije (E. coli, Salmonella spp., Serratia marcescens),rjeđe - Burkholderia cepaciai Nocardia farcinica.Karakterističan je razvoj jetrenih i subfreničnih apscesa, osteomielitisa, pararektalnih apscesa, sepse.
Najteža, po život opasna zarazna komplikacija je aspergiloza, koja se može javiti u obliku difuznog oštećenja pluća i drugih organa (masno tkivo, mozak, kosti, zglobovi, endokardij). Pacijenti s kroničnom granulomatoznom bolešću nakon cijepljenja BCG često razvijaju infekciju povezanu s cjepivom koja uključuje regionalne limfne čvorove. Lezije mikobakterija u bolesnika s kroničnom granulomatoznom bolešću mogu imati plućnu i izvanpulmonalnu lokalizaciju i dugotrajne su. Za bolesnike s kroničnom granulomatoznom bolešću karakteristično je zaostajanje u tjelesnom razvoju.
Liječenje.... Antimikrobna terapija: preventivna stalna primjena kotrimoksazola i antifungalnih lijekova (itrakonazol, itd.); u slučaju zaraznih komplikacija, kombinirana antibiotska terapija (2-3 baktericidna antibiotika koja prodiru unutarćelijski) u kombinaciji s antifungalnom terapijom provodi se parenteralno. S razvojem aspergiloze indicirana je dugotrajna primjena amfotericina B ili kaspofungina. S mikobakterijskom infekcijom koristi se kombinacija dugotrajne specifične terapije s lijekovima protiv tuberkuloze s antibioticima širokog spektra. ... Kirurško liječenje često je popraćeno suppuracijom postoperativne rane i stvaranjem novih žarišta. Moguća je punkcijska drenaža apscesa pod ultrazvučnom kontrolom. ... Za liječenje teških zaraznih komplikacija s neučinkovitošću antibiotske terapije moguće je koristiti granulocitnu masu, visoke doze IFNy i G-CSF. ... Transplantacija koštane srži ili transplantacija stanica krvi pupkovine od kompatibilne braće i sesta može biti uspješna u ranoj dobi kada je rizik od smrti od zaraznih komplikacija i bolesti presadka naspram domaćina minimalan.
Defekti adhezije leukocita
Do danas su opisana 3 oštećenja adhezije leukocita. Svi imaju
autosomno recesivno nasljeđivanje, karakterizirano ponavljajućim i kroničnim bakterijskim i gljivičnim zaraznim bolestima. Tip I karakterizira odsutnost ili smanjena ekspresija CD11 / CD18 na leukocitima, poremećena kemotaksija neutrofila, leukocitoza (više od 25x10 9), kasni gubitak pupkovine i razvoj omfalitisa, loše zacjeljivanje rane i odsutnost stvaranja gnoja na mjestu prodiranja patogena u tijelo.
Liječenje.... Antibiotska terapija: zarazne epizode i profilaktičke. ... U težim slučajevima propisana je transplantacija koštane srži od HLA-kompatibilnog davatelja.
NEDOSTACI KIT SUSTAVA
Bolesti s nedostatkom komponenata komplementa
Manifestacije genskih defekata pojedinih komponenata sustava komplementa prikazane su u tablici. 11-2.
Nasljedni AD.Rijetko se otkrivaju bolesti uzrokovane nedostatkom komponenata komplementa, jer je za njihovu manifestaciju neophodno homozigotno stanje za autosomne \u200b\u200balele. Uz C1inh (inhibitor C1 esteraze) povezana je jedna iznimka: genska mutacija C1inh,što dovodi do nedostatka inhibitora, u heterozigotnom se stanju očituje u fenotipu poznatom kao nasljedni AO (za više detalja vidi poglavlje 13, Angioedem).
Bolesti imunoloških kompleksa.Nedostatak C1-C4 očituje se razvojem bolesti imunoloških kompleksa - sistemskog vaskulitisa i oštećenja bubrega, što se zajednički naziva sistemski sindrom lupus eritematozusa (SLE).
Piogene infekcije.Nedostatak C3 (također čimbenici H i I) povezan je s povećanom osjetljivošću na piogene infekcije. Nedostaci komponenata uključenih u alternativni put aktivacije komplementa, kao i nedostaci komponenata C5-C8, povezani su s povećanom osjetljivošću na infekciju uzrokovanu Neisseria spp.Nedostatak C9 obično je klinički asimptomatski.
Tablica 11-2.Kliničke manifestacije nedostataka u pojedinim komponentama sustava komplementa
Nedostatak lektina koji veže manozu
Nedostatak lektina koji veže manozu (poznat i kao lektin koji veže manozu - MSL) uzrokovan je defektom gena MBL(razne točkaste mutacije i delecije u genu MBLotkriven u 17% ljudi kavkaske rase). S defektima gena, oštećena je aktivacija proteaza koje razgrađuju komponente komplementa C2 i C4 i aktivacija sustava komplementa duž lektinskog puta. Kliničkiova se patologija očituje zaraznim sindromom.
Podaci laboratorijskih istraživanja.Analiza subpopulacija limfocita, leukocita, kao i izotipova imunoglobulina ne pokazuje značajne abnormalnosti, adekvatne kliničkim simptomima. U serumu krvi nema MSL-a.
Liječenje.Ova bolest nije klasični poremećaj imunodeficijencije. Stoga je imunokorekcija imunotropnim sredstvima kontraindicirana. Rekombinantni MSL može se koristiti kao farmakološko sredstvo za etiopatogenetsku nadomjesnu terapiju u bolesnika s ovom nasljednom greškom. Ovaj lijek je trenutno u kliničkom ispitivanju.
Prolazna hipogamaglobulinemija u djece
Privremena hipogamaglobulinemija u djece povezana je s fiziološkim obilježjem faznog stvaranja imunoglobulinskog sustava. Sazrijevanje stvaranja IgM i IgA antitijela najviše se "odgađa". U zdrave djece sadržaj majčinih IgG postupno se smanjuje i nakon šest mjeseci povećava se proizvodnja vlastitih IgG antitijela. Međutim, kod neke djece porast razine imunoglobulina je odgođen. Takva djeca mogu patiti od ponavljajućih bakterijskih zaraznih bolesti. U tim se slučajevima ne smije pribjegavati infuzijama donornog imunoglobulina (intravenski imunoglobulin).
Selektivni nedostatak imunoglobulina A
Selektivni nedostatak imunoglobulina A (SD IgA - Selektivni nedostatak IgA)razvija se kao rezultat defekta gena tnfrsf13b
ili p). Nedostatak IgA u prisutnosti imunoglobulina drugih klasa najčešća je imunodeficijencija otkrivena u općoj populaciji s učestalošću 1: 500-1500 ljudi (još češće u bolesnika s alergijama). Razlikovati selektivni nedostatak IgA, t.j. koji se sastoji u nedostatku jedne od potklasa (30% slučajeva) i potpune (70% slučajeva). Nedostatak u podrazredu IgA2 dovodi do izraženije kliničke slike od nedostatka u podrazredu IgA1. Moguće su i kombinacije nedostatka IgA s drugim poremećajima: s nedostatkom u biosintezi IgG i s abnormalnostima T-limfocita. Velika većina ljudi s selektivnim
nedostatak IgA je praktički zdrav. Za djecu mlađu od 2 godine nedostatak IgA je fiziološko stanje.
Smanjenje koncentracije IgA u serumu na<5 мг/дл у детей старше 4 лет; IgG и IgM в норме, количество и соотношение субпопуляций лимфоцитов и их функциональная активность могут быть в норме.
Klinička slika.S nedostatkom IgA mogu se razviti 3 skupine patoloških sindroma: infektivni, autoimuni i alergijski. Pacijenti s nedostatkom IgA skloni su ponavljajućim infekcijama gornjih dišnih putova i probavnog sustava. Najčešće i najteže su razne autoimune bolesti (reumatoidni artritis, ankilozirajući spondilitis, Sjogrenov sindrom, vaskulitis s oštećenjem cerebralnih žila, autoimuni tiroiditis, SLE, glomerulonefritis, hemolitička anemija, dijabetes melitus tipa I, vitiligo itd.) Incidencija celijakije za 10 puta prelazi onu u djece s normalnim IgA. Najčešće otkrivene alergijske manifestacije: netolerancija na proteine \u200b\u200bkravljeg mlijeka, atopijski dermatitis (AD), bronhijalna astma.
Liječenje.Asimptomatski slučajevi ne zahtijevaju nikakav poseban tretman; u prisutnosti kliničkih manifestacija zaraznih, autoimunih i alergijskih bolesti, liječenje se provodi u skladu sa standardima.
Zamjenska terapija donornim imunoglobulinima nije indicirana ni kod selektivnog ni kod potpunog nedostatka IgA, jer postoji velika vjerojatnost stvaranja antiizotipskih protutijela na IgA u primatelja i razvoja transfuzijskih komplikacija koje oni uzrokuju.
Agamaglobulinemija s nedostatkom B stanica
X-vezana agamaglobulinemija (Brutonova bolest)čini 90% svih slučajeva agamaglobulinemije. Dječaci, sinovi (אּ, ρ) nositelja neispravnog gena su bolesni btk (Xq21,3-q22),kodirajući B-limfocite specifičnu protein tirozin kinazu Btk (Brutonova tirozin kinaza- Brutonova tirozin kinaza). Kao rezultat defekta, poremećeni su unutarstanični signalni putovi, rekombinirani su teški lanci imunoglobulina,
pretvaranje pre-B-stanica u B-limfocite. U 10% bolesnika agamaglobulinemija s nedostatkom B-stanica nasljeđuje se na autosomno recesivni način. Trenutno je opisano 6 genetskih defekata, uključujući receptor za B-stanice, citoplazmatski protein B-stanični adapter (BLNK) i gen Leucinom bogato ponavljanje 8 (LRRC8).
Podaci laboratorijskih istraživanja.Nema perifernih B limfocita. Koštana srž sadrži pre-B stanice s μ-lancem u citoplazmi. Broj T-limfocita i testovi funkcije T-limfocita mogu biti normalni. IgM i IgA u krvi se ne mogu otkriti; IgG može biti prisutan, ali u malim količinama (0,4-1,0 g / l). Ne postoje antitijela na antigene krvnih grupa i antigene cjepiva (tetanus, toksini difterije itd.). Može se razviti neutropenija. Histološki pregled limfoidnog tkiva: u limfoidnim folikulima nema zametnih (embrionalnih) centara i plazma stanica.
Klinička slika.Ako nije poznata obiteljska povijest, dijagnoza postaje očita u prosjeku za 3,5 godine. Bolest je karakterizirana hipoplazijom limfoidnog tkiva, teškim gnojnim infekcijama, zaraznim bolestima gornjeg (sinusitis, otitis media) i donjeg (bronhitis, upala pluća) respiratornog trakta; mogući gastroenteritis, piodermija, septički artritis (bakterijski ili klamidijski), septikemija, meningitis, encefalitis, osteomielitis. Uzročnici respiratornih bolesti su najčešće Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus,proljev crijevne bakterije ili lamblije Giardia lamblia.Također, bolesnici s agamaglobulinemijom skloni su zaraznim bolestima uzrokovanim mikoplazmama i ureaplazmama, koje su uzrok razvoja kronične upale pluća, gnojnog artritisa, cistitisa i apscesa potkožnog tkiva. Tipični virusi su neurotropni virusi ECHO-19 i coxsackie, koji uzrokuju teški akutni i kronični encefalitis i encefalomielitis. Manifestacije enterovirusnih infekcija mogu biti sindrom sličan dermatomiozitisu, ataksija, glavobolje i poremećaji u ponašanju. U bolesne djece, kada se imunizira živim cjepivom protiv dječje paralize, u pravilu se otkriva produljeno prolijevanje virusa poliomijelitisa kroz sluznicu te s obnovljenom i sve većom virulencijom (tj.
postoji stvarna opasnost od zaraze zdrave djece poliomijelitisom kao rezultat kontakta s cijepljenim imunodeficijentnim djetetom). Autoimune poremećaje u agamaglobulinemiji mogu predstavljati reumatoidni artritis, sindrom sličan sklerodermu, skleredem, ulcerozni kolitis, dijabetes melitus tipa I (zbog prevladavanja imunološkog odgovora Th1).
Sistematski pregled.Obratite pažnju na zaostajanje u tjelesnom razvoju, na oblik prstiju (prsti u obliku bataka), promjene oblika prsnog koša, karakteristične za bolesti donjih dišnih putova, hipoplaziju limfnih čvorova i tonzila.
Liječenje.
Supstitucijska terapija: intravenski pripravci imunoglobulina primjenjuju se svaka 3-4 tjedna tijekom života. Doze imunoglobulina odabiru se tako da stvore koncentraciju u serumu pacijenta koja preklapa donju granicu dobne norme.
Razgovarajte o mogućnosti genske terapije - gen Btkklonirano, ali njegova prekomjerna ekspresija povezana je sa zloćudnom transformacijom krvotvornog tkiva.
U slučaju trajne neutropenije, koriste se faktori rasta. Kada se pojave znakovi autoimune patologije, moguće je propisati monoklonska antitijela (infliksimab, itd.).
Uobičajeni varijabilni imunološki nedostatak
Općenito varijabilni imunološki nedostatak (CVID) skupina je sindroma koju karakterizira nedostatak u sintezi antitijela i staničnom imunitetu. Pouzdan dijagnostički kriterij za CVID je značajno smanjenje sadržaja imunoglobulina dva ili tri glavna izotipa u oba spola u kombinaciji s jednim od sljedećih znakova:
Početak bolesti u dobi starijoj od 2 godine;
Nedostatak izohemaglutinina i / ili nizak odgovor na cijepljenje;
Isključenje drugih uzroka agamaglobulinemije.
U nekih su bolesnika uzrok razvoja CVID mutacije gena koji kodiraju molekule uključene u procese sazrijevanja i preživljavanja B stanica: BAFF-R (Receptor za aktiviranje faktora B-stanica),Blimp-1 (B-limfociti izazvani sazrijevanjem protein-1)i ICOS (Inducibilni kostimulator).Dolazi do kršenja sposobnosti B-limfocita da se diferenciraju u plazma stanice, razvijaju se nedostaci u stvaranju antitijela, moguća je disfunkcija T-limfocita i uočava se povećana sklonost zaraznim bolestima. Sindrom se može manifestirati u ranom djetinjstvu, adolescenciji ili mladim odraslima.
Podaci laboratorijskih istraživanja.Razine IgG i IgA (u oko 50% bolesnika) i IgM (do neotkrivenih količina) značajno su smanjene. Broj B-limfocita u krvi je normalan ili smanjen. Broj T-limfocita u većine je bolesnika normalan. U ozbiljnih bolesnika može se razviti limfopenija (manje od 1500x103 stanica u 1 L krvi). Broj NK stanica je smanjen. Proizvodnja specifičnih antitijela kao odgovor na imunizaciju smanjena je ili odsutna. Proliferacija limfocita i stvaranje IL-2 pod utjecajem mitogena i antigena znatno su oštećeni.
Klinička slika.Otkrivaju se ponavljajuće bakterijske zarazne bolesti s lokalizacijom uglavnom u respiratornom traktu i paranazalnim sinusima. U vrijeme dijagnoze infekcije respiratornog trakta mogu preći u bronhiektazije i difuzne lezije plućnog tkiva. Moguća je zarazna lezija probavnog sustava, koja se očituje proljevom, steatorejom i malapsorpcijom (i, shodno tome, gubitkom težine). Infekcije uzrokovane Giardia lamblia, Pneumocystis cariniiili obiteljske viruse Herpetoviridae.Pacijenti s CVID-om skloni su razvoju gnojnog artritisa uzrokovanog mikoplazmama i ureaplazmama. Manifestacije enterovirusnih infekcija mogu biti encefalomielitis, poliomijelitis i sindromi slični dermatomiozitisu, lezije kože i sluznice. Autoimunebolesti su teške i mogu odrediti prognozu CVID-a. Ponekad su prve kliničke manifestacije CVID artritis, ulcerozni kolitis i Crohnova bolest, sklerozirajući holangitis, malapsorpcija, SLE, nefritis, miozitis, autoimuna bolest pluća u obliku limfoidnog intersticijskog pneumonitisa, neutropenija,
trombocitopenična purpura, hemolitička anemija, perniciozna anemija, alopecija totalis, vaskulitis mrežnice, fotosenzibilnost. Bolesnici s CVID-om imaju značajno povećanu učestalost maligne novotvorine(u 15% slučajeva), sarkoidozi slični granulomi i nemaligna limfoproliferacija. Liječenje.
Antibakterijska kemoterapija.
Supstitucijska terapija: intravenski pripravci imunoglobulina primjenjuju se svaka 3-4 tjedna tijekom života.
U slučaju autoimunih komplikacija - imunosupresivna terapija (glukokortikoidi, azatioprin, ciklosporin A) i eventualno imenovanje monoklonskih antitijela (infliksimab, itd.).
Hiper-IgM sindromi
Hiper-IgM sindromi prilično su rijetke bolesti koje karakterizira izraženo smanjenje ili potpuno odsustvo IgG, IgA i normalna ili povećana koncentracija IgM u serumu. To je zbog nemogućnosti B-limfocita da prebace klase imunoglobulina i hipermutageneze varijabilnih domena. Do danas je identificirano 6 genetskih nedostataka koji dovode do razvoja hiper-IgM sindroma.
. Tip 1 (HIGM 1).X-povezani nedostatak CD40 liganda (70% slučajeva hiper-IgM sindroma), što dovodi do nesposobnosti T stanica da učinkovito komuniciraju s B limfocitima.
. Tip 2 (HIGM 2).Autosomno recesivno, povezano s aktivacijom citidin deaminaze izazvanom AID defektom (gen Aicda, 12p13)- enzim koji sudjeluje u izmjeni klasa imunoglobulina i hipermutagenezi.
. Tip 3 (HIGM 3).Autosomno recesivno, povezano s genskom mutacijom molekule CD40. U ovom slučaju, B stanice same po sebi nisu u mogućnosti učinkovito komunicirati s T limfocitima. Fenotipske manifestacije slične su onima tipa 1.
. Tip 4 (HIGM 4).Autosomno recesivno; u nekim se slučajevima javljaju mutacije de novo.Povezan s defektom u UNG - uracil-DNA glikozilaza - enzim koji je također uključen u
u zamjenjivanju klasa imunoglobulina, ali nakon djelovanja AID-a. U ovom slučaju, hipermutageneza nije pogođena i sindrom teče s manje ozbiljnosti.
. Tip 5 (HIGM 5).Oštećena samo prebacivanjem klase, hipermutageneza nije pogođena. Uzročna mutacija još nije utvrđena, ali očito postoji nedostatak u enzimu koji djeluje nakon toga
. Tip 6 (HIGM-ED).X-povezano, povezano s dishidrotskom ektodermalnom displazijom, uzrokovano je nedostatkom NEMO (NF-kB modulatora), što rezultira oslabljenom CD40 signalizacijom.
X-povezani hiper-IgM sindromidentificirani češće od ostalih. Razvija se s nedostatkom u genu koji kodira CD40L (CD154, gen je smješten na Xq26-q27.2)je ligand za CD40. Nedovoljna ekspresija CD40L od strane T-limfocita dovodi do nemogućnosti prebacivanja klase imunoglobulina u B-limfocitima s IgM na druge izotipe, kao i do poremećenog stvaranja memorijskih B-stanica, repertoara T-stanica i odgovora Th1-stanica usmjerenih protiv unutarstaničnih mikroorganizama. Dječaci su bolesni
Podaci laboratorijskih istraživanja.IgG, IgA, IgE nije moguće odrediti ili se otkrivaju u vrlo malim količinama. Razine IgM su normalne (u 50% slučajeva) ili povišene, često značajno. Broj T i B stanica je normalan; smanjeni proliferativni odgovor T stanica, induciran antigenima. IgM su poliklonski, ponekad i monoklonski. Otkriti autoantitijela izotipa IgM (anti-eritrociti, antitrombociti, antitiroidni, antitijela na antigene glatkog mišićnog tkiva). U limfoidnom tkivu nema zametnih centara, ali postoje plazma stanice.
Klinička slika.Prve manifestacije javljaju se u djetinjstvu i ranom djetinjstvu. Ponovljeno infekcijerazličita lokalizacija (prvenstveno dišni put), uključujući oportunističku (uzrokovanu Pneumocystis carinii).Također su karakteristične lezije virusima (citomegalovirus i adenovirusi), Criptococcus neoformans,mikoplazme i mikobakterije. Kriptosporidijalna infekcija može uzrokovati akutni i kronični proljev (razvija se u 50% bolesnika) i sklerozirajući kolangitis. Anemija, neutropenija, ulceracija usne sluznice, gingivitis, ulcerativni
lezije jednjaka, razni dijelovi crijeva, ulcerozni kolitis. Otkrivaju sklonost ka autoimuni poremećaji(seronegativni artritis, glomerulonefritis, itd.) i maligne novotvorine (uglavnom limfoidno tkivo, jetra i žučni trakt). Mogu se razviti limfadenopatija, hepato- i splenomegalija. Liječenje
Redovita nadomjesna terapija intravenskim imunoglobulinom.
Antibakterijska kemoterapija. Za prevenciju i liječenje pneumocystis upale pluća koriste se ko-trimoksazol [sulfametoksazol + trimetoprim] i pentamidin.
Da biste spriječili oštećenje jetre i žučnih puteva, trebali biste koristiti samo prokuhanu ili filtriranu vodu, provoditi redovite preglede (ultrazvuk, biopsija jetre ako je naznačeno).
U liječenju neutropenije i oralne ulceracije koriste se glukokortikoidi i pripravci koji stimuliraju koloniju granulocita.
S razvojem autoimunih komplikacija propisana je imunosupresivna terapija (glukokortikoidi, azatioprin, ciklosporin A), kao i lijekovi na bazi monoklonskih antitijela.
Optimalna metoda liječenja je transplantacija koštane srži od davatelja koji se podudaraju s HLA-om (stopa preživljavanja 68%, najbolje učiniti prije 8. godine).
Ovo je najčešća primarna imunodeficijencija susreće se s frekvencijom 1/300 - 1/700. Prvi put je ...
opisao J. Heremans 1960.
U većini slučajeva ne uočava se jasna slika nasljeđivanja bolesti; u rjeđim slučajevima obiteljskog gubitka IgA, autosomno recesivno nasljeđivanje javlja se češće od dominantnog nasljeđivanja. Nedostatak IgA ponekad je povezan s abnormalnostima na 18. kromosomu.
IgA se razlikuje od ostalih imunoglobulina po sadržaju ugljikohidrata i sijaličnih kiselina i sposobnosti stvaranja dimera, trimera, pa čak i tetramera. Postoje dvije varijante molekula IgA:
- serumski IgA je uvijek monomer;
- sekretorni IgA - tvori di-, tri- i tetramere prije nego što postane sastavni dio sekreta (slina, suze, mlijeko, sekreti probavnog, dišnog i mokraćnog sustava).
Poseban proteinski lanac, J-lanac, sudjeluje u stvaranju sekretornog IgA. Stoga je ovo čitav sustav bjelančevina. Stoga - oblici patologije IgA:
- Općeniti nedostatak IgA povezan je s abnormalnostima u sintezi IgA monomera. Kao rezultat, smanjuje se sadržaj i seruma i sekretornog IgA. Krši se i lokalna i opća zaštita;
- kvar u stvaranju sekretornih molekula - SIgA. Razlog može biti odsutnost J-lanca, što dovodi do kršenja lokalnog imuniteta;
- pretežno kršenje sinteze serumskog IgA, kada se oslobađaju samo sekretorni oblici IgA, a serumski IgA monomeri praktički ne izlučuju.
Klinički simptomi ovise o nedostatak IgA a može se manifestirati kao patologija pojedinih organa i sustava. Infektivne lezije sluznice su najčešće.
Iz gastrointestinalnog trakta - ovo je kronični gastritis, ulcerozni i hemoragični kolitis, ileitis, stomatitis. Dio dišnog sustava - rinitis, sinusitis, bronhitis, koji brzo postaju kronični. Kronična bronhopneumonija s ishodom u bronhiektazijama i apscesima pluća je česta. Također je utvrđena povezanost između smanjenog sadržaja sekretornog IgA i povećane učestalosti atopijske alergije i razvoja autoimunih bolesti (sistemski eritemski lupus, reumatoidni artritis, autoimuni tiroiditis).
Pojedinci s selektivnim nedostatkom pretežno monomernog IgA u serumu najčešće nemaju kliničke manifestacije imunodeficijencije.
Laboratorijska istraživanja:pokazuju da je ukupan broj T- i B-limfocita u perifernoj krvi kod takvih bolesnika u granicama normale. Razina seruma i / ili sekretornog IgA naglo je smanjena, dok su razine IgM i IgG u normalnim granicama. U nekih bolesnika (s manifestacijama atopijske alergije) razina IgE može se povećati.
Liječenje: simptomatski. Pri upotrebi nadomjesne terapije imunoglobulinom mora biti oprezan, jer ponovljenim davanjem postoji rizik od anafilaktičkog šoka zbog mogućnosti povećane proizvodnje anti-imunoglobulina klase IgG i IgM.
Prognoza: tijek bolesti je uglavnom dobroćudan.
Dijagnostički kriterij - smanjenje u bolesnika starijih od 4 godine razine serumskog IgA manje od 0,07 g / l s normalnim razinama IgG i IgM i isključenje drugih uzroka hipogamaglobulinemije. Dijagnostički značajno:
Izolirano smanjenje razine IgA u serumu (manje od 0,05 g / l) s normalnim razinama ostalih izotipova imunoglobulina u djece starije od 1 godine, odsutnost IgAl i IgA2. Razine IgM i IgG su normalne. Međutim, neki pacijenti imaju nedostatak IgG2;
- ako je razina IgA u rasponu od 0,05 g / l do 0,2 g / l, tada se dijagnosticira djelomični nedostatak IgA; normalan broj T-limfocita i njihovih podrazreda;
- obično normalan broj B-limfocita (CD19 \\ CD20);
- normalan broj NK stanica (CD16 CD56).
U bolesnika s nedostatkom IgA, posebno u nedostatku sekretornog IgA, treba ispitati razinu podrazreda IgA. U nekih bolesnika, selektivni nedostatak IgA može u budućnosti napredovati razvojem CVID-a. Potrebno je dugoročno redovito praćenje sadržaja imunoglobulina (uključujući i kod asimptomatskih bolesnika).
Određivanje autoantitijela (antinuklearna, antitiroidna, itd.).
S hranom netolerancija ili malapsorpcija zahtijeva testiranje na alergiju i određivanje antitijela na mlijeko i antiglutenska IgG antitijela.
Liječenje... Pacijenti s asimptomatskim selektivnim nedostatkom IgA ne zahtijevaju kontinuirano liječenje. Pacijentima s manifestacijama zaraznih bolesti u profilaktičke svrhe propisani su antibiotici. Intenzivno antibakterijsko liječenje provodi se kod svih bolesnika tijekom pojave zarazne bolesti. Rutinska imunizacija nije kontraindicirana u bolesnika. Zamjenska terapija imunoglobulinom kontraindicirana je kada se u pacijenta otkriju anti-IgA autoantitijela. Treba imati na umu da se selektivni nedostatak IgA odnosi na neispravljene primarne imunološke nedostatke. Terapijske mjere svode se na simptomatsku terapiju zaraznih, alergijskih i autoimunih bolesti. Imunotropni lijekovi propisani su uglavnom u vezi s manifestacijom povećanog zaraznog morbiditeta.
Prognoza... U bolesnika s selektivnim nedostatkom IgA, prognoza ovisi o prisutnosti popratnih nedostataka u određenim antitijelima, alergijama ili autoimunim bolestima. Često se asimptomatski tijek bolesti može poremetiti zbog djelovanja vanjskih štetnih čimbenika, na primjer, u stresnoj situaciji, imunosupresijom, kemoterapijom itd.