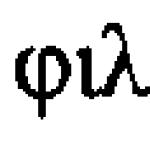Ar trebui diferențiat de sindromul real al hipervitaminozei D, în care, ca urmare a deficienței enzimatice a aparatului tubular renal, severă tulburări metaboliceîn corpul copilului. V. V. Shitskova et al. (1971), pe baza propriilor observații, furnizează următorul tabel de diagnostic diferențial al acestor boli (Tabelul 7) cu unele dintre completările noastre.
| Indicatori | Hipervitaminoza D | Sindromul De Toni-Debreu-Fanconi |
| Frecvenţă | Relativ des | Rareori |
| Patogeneza | Încălcare procesele metabolice, în principal calciu, din cauza unei supradoze de vitamina D | Enzimopatie. Tubulopatie congenitală. Reabsorbția afectată a fosforului, glucozei și azotului amino |
| Tabloul clinic | Piele uscată și palidă, sete, vărsături, constipație, malnutriție, hipertensiune arterială, mărire a ficatului | Piele uscată și palidă, anorexie, sete, vărsături, constipație, poliurie, malnutriție, mărire a ficatului. Fara hipertensiune arteriala. Hipotonia mușchilor |
| Teste biochimice de sânge | Hipercalcemia în perioada acuta. Fosforul este scăzut. Zaharul si proteinele sunt normale. Fosfataza alcalină nu este modificată | Calciul este normal sau redus. Fosforul este redus brusc. Zaharul și proteinele sunt reduse, activitatea fosfatazei alcaline este puternic crescută. Acidoza metabolica |
| Urină | Reacția lui Sulkowicz este pozitivă. Proteinurie, microhematurie, leucociturie. Zahărul cu azot amino este adesea normal | Reacția lui Sulkowicz este negativă. Proteinurie, fosfaturie, glucozurie, aminoacidurie |
| Radiografia oaselor tubulare | Extinderea și compactarea zonelor de pre-calcificare | Osteoporoza oaselor lungi, zonele de calcificare sunt sărace |
sindromul Fanconi- diabet cu amine. Tulburarea metabolismului cistinei însoțită de glicozurie, aminoacidourie, fosfaturie, secretie crescuta alanină, pierdere de alcalii. Cauzat de un defect ereditar al tubilor renali, determinând incapacitatea de a reabsorbi glucoza, aminoacizii și fosfatul. Metabolismul cistinei este perturbat, care se depune sub formă de cristale în sistemul reticuloendotelial, cornee, tubuli renali și alte țesuturi. Acest lucru duce la eșec progresiv în timp diverse funcții tubii renali.
Sindromul Fanconi trebuie distins de cistinurie, în care cistina nu se depune în țesuturi. Odată cu sindromul, capacitatea funcțională a aparatului glomerular rămâne normală. Concentrația de calciu seric este normală, nivelul de fosfor anorganic scade brusc. Radiografia arată osteomalacia și epuizarea difuză a oaselor în alcali.
Sindromul Fanconi (de Toni-Debreu-Fanconi) este considerat o disfuncție tubulară „majoră”, caracterizată prin reabsorbție afectată a majorității substanțelor și ionilor (aminoacidurie, glicozurie, hiperfosfaturie, excreție crescută de bicarbonat) și modificări metabolice sistemice.
Sindromul Fanconi implică multiple defecte de reabsorbție la nivelul tubilor renali proximali, ducând la glicozurie, fosfaturie, aminoacidurie generalizată și scăderea concentrației de bicarbonat. Simptomele la copii includ malnutriție, întârziată dezvoltare fizică si rahitism, simptome la adulti - osteomalacie si slăbiciune musculară. Diagnosticul se bazează pe detectarea glicozuriei, fosfaturiei și aminoaciduriei. Tratamentul include compensarea deficitului de bicarbonat, precum și tratamentul insuficienta renala.
Cod ICD-10
E72.0 Tulburare de transport de aminoacizi
Epidemiologie
Sindromul Fanconi apare în diferite regiuni ale lumii. Incidența bolii, conform datelor moderne, este de 1 la 350.000 de nou-născuți. Aparent, nu se ia în calcul doar sindromul Fanconi, ci și sindromul Fanconi care s-a dezvoltat în perioada nou-născutului.
Cauzele sindromului Fanconi
Sindromul Fanconi este congenital sau se dezvoltă ca parte a unor boli dobândite.
Natura defectului genetic și produsul biochimic primar rămân prost înțelese. Se crede că baza este fie o anomalie a proteinelor de transport ale tubilor renali, fie o mutație genică care asigură inferioritatea enzimelor care determină reabsorbția glucozei, aminoacizilor și fosforului. Există dovezi ale tulburărilor mitocondriale primare în sindromul Fanconi. Defectul genetic determină severitatea bolii. Există sindromul Fanconi complet și incomplet, adică pot exista toate cele 3 defecte biochimice principale sau doar 2 dintre ele.
Factori de risc
Sindromul Fanconi (boala de-Toni-Debreu-Fanconi) este considerat mai des ca un sindrom asociat cu cistinoză, galactozemie, glicogenoză, tirozinemie, intoleranță la fructoză, boala Konovalov-Wilson, leucodistrofie metacromatică, deficit de piruvat carboxilază, expunere deficit de carboxilază mitocondrivă la substanțe toxice da, aminoglicozide, tetracicline expirate, metale grele) sau în curs de dezvoltare în legătură cu boli dobândite precum amiloidoza, deficitul de vitamina D etc. Cu toate acestea, după o serie de autori, sindromul Fanconi poate fi o boală independentă, una dintre cele mai severe boli asemănătoare rahitismului.
Patogeneza
În literatura de specialitate, termenul „sindrom Fanconi” sau „sindrom Debra de Toni-Fanconi” este mai des utilizat termenii „diabet glucoaminofosfat”, „diabet cu glucofosfamină”, „nanism renal cu rahitism rezistent la vitamina D”, „idiopatic; sindrom renal” sunt de asemenea frecvente Fanconi”, „. sindrom ereditar Fanconi.” Termenii cei mai des întâlniți în literatura străină sunt: „sindromul Fanconi renal”, „sindromul Fanconi”, „sindromul de-Tbni-Debre-Fanconi primar”, „sindromul Fanconi moștenit” etc.
Datele clinice și experimentale susțin întreruperea transportului transmembranar în tubul contort proximal al nefronului. Încă nu a fost clarificat dacă un defect structural sau biochimic stă la baza bolii. Modificări asemănătoare rahitismului se dezvoltă fie datorită influenței combinate a acidozei și hipofosfatemiei, fie numai hipofosfatemiei. Potrivit unui număr de cercetători, patologia se bazează pe o scădere a rezervelor intracelulare de ATP.
Sindromul Fanconi ereditar de obicei îi însoțește pe alții boli congenitale, în special cistinoză. Sindromul Fanconi poate fi, de asemenea, asociat cu boala Wilson, intoleranța ereditară la fructoză, galactozemie, boli de stocare a glicogenului, sindromul Lowe și tirozinemia. Modelul de moștenire variază în funcție de boala asociată.
Sindromul Fanconi dobândit poate fi cauzată de diverse medicamentele, inclusiv unele medicamente pentru chimioterapie anticancer (de exemplu, ifosfamidă, streptozocină), antiretrovirale (de exemplu, didanozină, cidofovir) și tetraciclină expirată. Toate aceste medicamente sunt nefrotoxice. Sindromul Fanconi se poate dezvolta și cu transplant de rinichi, mielom multiplu, amiloidoză, intoxicație cu metale grele și alți agenți chimici sau deficit de vitamina D.
Simptomele sindromului Fanconi
Simptomele sindromului Fanconi sunt variate. La copii, simptomele seamănă mai des cu diabetul fosfat. La adulți, se observă poliurie, hipostenurie, slăbiciune musculară și dureri osoase. Posibil hipertensiune arterială, în absența tratamentului - formarea insuficienței renale cronice.
De regulă, primele manifestări ale bolii apar în primul an de viață al unui copil. Adevărat, la cei 10 copii pe care i-am observat cu boala pre-Toni-Debreu-Fanconi, primele simptome au apărut după un an și jumătate de viață. În primul rând, poliuria și polidipsia, febra de grad scăzut, vărsăturile și constipația persistentă atrag atenția. Copilul începe să rămână în urmă în dezvoltarea fizică, deformările osoase apar în principal membrele inferioare tip valgus sau varus. Se dezvoltă hipotonia musculară, iar la vârsta de 5-6 ani copiii nu pot merge independent. Odată cu progresia tulburărilor tubulare cu 10-12 ani de viață, este posibilă dezvoltarea insuficienței renale cronice. Pe lângă simptomele de mai sus, modificări patologiceși din alte organe. Dintre cei 10 copii menționați mai sus care se aflau sub supravegherea noastră, 7 aveau anomalii oftalmologice, 6 aveau patologie a sistemului nervos central, 5 aveau patologie. sistemul cardiovascularși anomalii anatomice ale organelor sistemul urinar, în 4 - patologia organelor ORL și a tractului gastrointestinal, în cazuri izolate - tulburări endocrineși stări de imunodeficiență.
Diagnosticul sindromului Fanconi
Pentru a confirma diagnosticul, studii de contrast cu raze X ale oaselor și extinse teste de laborator sânge și urină.
Diagnosticul de laborator al sindromului Fanconi
ÎN analiză biochimică sânge trăsături caracteristice ia în considerare o scădere a conținutului de calciu (2-microglobuline. Ei observă o scădere a concentrației de sodiu și potasiu în sânge, o creștere a clearance-ului acid uric cu scăderea conținutului său în sânge. Pierderea excesivă de bicarbonat în urină duce la o imagine pronunțată a acidozei metabolice. O încălcare a bioenergeticii a fost dezvăluită sub forma unei scăderi a activității enzimatice metabolismul energetic: a-glicerofosfat dehidrogenază, glutamat dehidrogenază, succinat dehidrogenază. În același timp, aproape toți pacienții au prezentat o încălcare a peroxidării sub forma unei creșteri a conținutului de acizi lactic și piruvic din sânge.
Teste de laborator
- Aminoacidurie generalizată.
- Acidoză tubulară renală proximală cu bicarbonaturie.
- Fosfaturie, hipofosfatemie, diabet fosfatic.
- Hiposhenurie, poliurie.
- Proteinurie tubulară (beta 2 microglobuline, lanțuri ușoare de imunoglobuline, proteine cu greutate moleculară mică).
- Hipokaliemie.
- Hipocalcemie.
- Hiponatremie.
- Hiperuricozurie.
Diagnosticul instrumental al sindromului Fanconi
Ca studii instrumentale obligatorii în diagnosticul sindromului Fanconi, radiografia oaselor scheletice este utilizată pe scară largă pentru a detecta deformările membrelor și tulburările structurale. țesut osos- osteoporoză (de obicei sistemică) și o întârziere a ratei de creștere a țesutului osos de la vârsta calendaristică a copilului. Țesutul osos este caracterizat de o structură fibroasă grosieră, iar epifizioliza este adesea detectată. ÎN secțiuni distale Tibia femurală și proximală prezintă o structură celulară de țesut osos și formațiuni asemănătoare pintenilor. În etapele ulterioare ale bolii, este detectată osteoporoza și sunt posibile fracturi osoase tubulare. Pentru a determina severitatea osteoporozei, se utilizează densitometria cu raze X.
La cercetarea radioizotopilor detectează acumularea unui radioizotop în zonele osoase de creștere intensivă a pacientului.
O examinare morfologică a biopsiilor de țesut osos arată o structură perturbată a fasciculelor osoase, dezvăluind lacune și mineralizare osoasă slabă.
În timpul nefrobiopsiei, se observă o imagine particulară a tubilor proximali (forma seamănă cu un „gât de lebădă”), se dezvăluie atrofia epitelială și fibroza interstițială. Glomerulii sunt implicați în proces chiar în etapele finale ale bolii. Examenul microscopic electronic evidențiază în epiteliu număr mare mitocondriile.
Exemple de formulare a diagnosticului
sindromul Fanconi. OMIM-134 600. Insuficiență renală cronică, stadiu terminal. Hiperparatiroidism secundar. Osteoporoza sistemica. Deformarea varusă a membrelor.
Glicogenoza tip I. sindromul Fanconi. Insuficiență renală cronică stadiul I.
Diagnostic diferenţial
Diagnosticul diferențial se realizează cu toate bolile în care se dezvoltă sindromul Fanconi. Acestea includ următoarele boli ereditare:
- galactozemie;
- glicogenoza tip I;
- tirozinemia;
- cistinoză;
- osteogeneza imperfectă;
- boala Konovalov-Wilson;
- talasemie;
- sindrom nefrotic congenital;
- acidoza tubulara renala.
Pe lângă bolile ereditare diagnostic diferenţial efectuate cu afecțiuni patologice dobândite:
- intoxicație cu metale grele, chimicaleŞi medicamente, mai ales cele expirate;
- hiperparatiroidism secundar;
- arsuri severe;
- mielom multiplu;
- diabet zaharat
Tratamentul sindromului Fanconi
Tratamentul sindromului Fanconi are ca scop corectarea hipokaliemiei, acidozei tubulare renale proximale și a altor tulburări electrolitice. Terapia diabetului cu fosfat se efectuează conform reguli generale. Pacienții cu sindrom Fanconi trebuie sfătuiți să bea multe lichide.
Cu sindromul Fanconi secundar, simptomele sale scad sau dispar complet odată cu tratamentul cu succes al bolii de bază.
Obiectivele tratamentului
Non-drog și tratament medicamentos pacienții cu boala Fanconi sunt foarte apropiați în esență, deoarece implică corectarea tulburărilor electrolitice (eliminarea deficitului de potasiu și bicarbonat), modificări ale echilibrului acido-bazic. Terapia simptomatică este, de asemenea, necesară.
Dietoterapia
Deoarece este necesar să se limiteze excreția aminoacizilor care conțin sulf, alimentele din cartofi și varză sunt potrivite ca agenți dietetici. Tratament medicamente active Suplimentarea cu vitamina D ar trebui efectuată pe o dietă cu sare limitată și includerea de produse care au efect alcalinizant: lapte, sucuri de fructe. Este necesar să se utilizeze pe scară largă preparate care conțin potasiu, caise uscate și stafide. În cazurile de deficit sever de potasiu, se recomandă adăugarea de panangin sau asparkam. Dacă acidoza este pronunțată, atunci dieta singură nu este suficientă;
Boala De Toni-Debreu-Fanconi (glucoaminofosfat-diabet) este cea mai gravă boală dintre R. b. Se moștenește în mod autosomal recesiv, dar expresivitatea genei mutante în starea homozigotă variază semnificativ; Există cazuri sporadice cauzate de o nouă mutație.
Se crede că în centrul bolii defecte determinate genetic în fosforilarea enzimatică în tubii renali (tubulopatie combinată). Acest lucru duce la o întrerupere a proceselor de aprovizionare cu energie pentru transportul fosfaților, glucozei și aminoacizilor în tubii renali și la creșterea excreției lor în urină, precum și la o tulburare a mecanismelor de menținere a echilibrului acizilor și bazelor. Dezvoltarea acidozei metabolice și a lipsei de compuși ai fosforului contribuie la perturbarea formării țesutului osos, cum ar fi osteomalacia și modificări asemănătoare rahitismului la nivelul scheletului. În unele cazuri este dezvăluit modificări morfologiceîn tubii renali, disfuncții ale glandelor paratiroide, tulburări ale sintezei 1,25-dioxicolecalciferolului.
În majoritatea cazurilor primul semne de boală apar în a doua jumătate a primului an de viață, un complex de simptome detaliat este format în al doilea an de viață; Manifestarea tardivă a bolii este mai puțin frecventă - la vârsta de 6-7 ani. Manifestările clinice inițiale sunt sete crescută, poliurie, uneori febră de grad scăzut prelungit, vărsături. În al doilea an de viață, se depistează dezvoltarea fizică întârziată și deformări osoase ale extremităților inferioare (valgus sau varus). piept, antebrațe și humerus(Fig. 3). Din punct de vedere radiologic, se determină osteoporoza sistemică de diferite grade de severitate, subțierea stratului cortical al oaselor tubulare, slăbirea zonelor de creștere și o întârziere a ratei de creștere a țesutului osos de la vârsta biologică a copilului.
Caracteristici caracteristice tulburările biochimice din boala de Toni-Debreu-Fanconi sunt scăderea nivelului de calciu și fosfor din sânge, creșterea activității fosfatazei alcaline, acidoza metabolică (pH - 7,35-7,25; BE = -10-12 mmol/). l). Excreția urinară de calciu rămâne de obicei normală cu clearance-ul urinar crescut al fosfatului. Se remarcă glucozurie (20-30 g/l și peste), hiperaminoacidurie generalizată și disfuncție a acidogenezei amoniacului - o scădere a acidității de titrare, o creștere a pH-ului urinei > 6,0. Cu poliurie de până la 2 litri sau mai mult pe zi, greutatea specifică a urinei este de obicei mare (1025-1035), care este de obicei asociată cu glucozurie.
În funcție de severitatea manifestărilor clinice și a tulburărilor metabolice, există două opțiuni clinice și biochimice boala De Toni-Debrau-Fanconi. Primul se caracterizează printr-o întârziere semnificativă a dezvoltării fizice, o evoluție severă a bolii cu pronunțată deformări osoaseși adesea fracturi osoase, hipocalcemie severă (1,6-1,8 mmol/l), scăderea absorbției de calciu în intestin. În cea de-a doua opțiune, se observă o întârziere moderată a dezvoltării fizice, o evoluție ușoară cu deformări osoase minore, normocalcemie și absorbție normală a calciului în intestin.
Criterii de diagnostic sunt o deficiență pronunțată a greutății și înălțimii corpului copilului, o întârziere în dezvoltarea funcțiilor statice-motorii, deformări ale scheletului asemănătoare rahitismului, cu o imagine caracteristică cu raze X a tulburărilor structurii țesutului osos și caracteristici observate ale tulburărilor electrolitice. .
Diagnostic diferenţial efectuat cu rahitism, osteopatii datorate insuficientei renale cronice; In plus, mare valoare are diferențierea bolii primare de Toni-Debreu-Fanconi cu sindromul secundar întâlnit în alte boli ereditare și dobândite (sindrom Low, nefronoftiză juvenilă, cistinoză, tirozinemia, galactozemie, glicogenoză, intoleranță ereditară la fructoză, distrofia hepatocerebrală, mieloma hepatocerebrală, distrofia mieloide, distrofia hepatocerebrală, distrofia mioidoză). sindrom nefrotic, transplant renal, hiperparatiroidism, afectarea rinichilor de la săruri de metale grele, otrăvire cu medicamente, inclusiv vitamina D, Lysol etc.). Sindromul Lowe (sindromul oculocerebrorenal), spre deosebire de boala De Toni-Debreu-Fanconi, se caracterizează printr-un decalaj în dezvoltare mentală, cataractă bilaterală, glaucom, hiporeflexie, precum și un tip de moștenire recesiv, legat de sex. Nefronoftiza juvenilă Fanconi, a cărei bază morfologică este chisturile medularei renale la nivelul canalelor colectoare și hialinoza glomerulilor renali, se caracterizează prin afectarea precoce a funcției de concentrare a rinichilor (hiposthenurie), anemie normocromă, hiperazotemie; modificările scheletice asemănătoare rahitismului apar mai târziu. Un studiu al biopsiei de țesut renal joacă un rol decisiv în diagnosticul nefronoftizei Fanconi.
Principii de bază tratament constau in corectarea tulburarilor electrolitice, modificarilor echilibrului acido-bazic, eliminarea deficitului de potasiu si bicarbonat. Caracteristicile utilizării vitaminei D și a metaboliților săi pentru a elimina tulburările homeostaziei fosfor-calcului se reduc la necesitatea unor cure repetate de tratament (doza zilnică inițială de vitamina D 25.000-30.000 unități, maxim - 75.000-150.000 unități, doza de oxidevit). 0,5-1,5 mcg pe zi), deoarece Când medicamentele sunt întrerupte, se observă adesea recăderi (așa-numitele crize metabolice, progresia osteoporozei și modificări asemănătoare rahitismului în țesutul osos). Complexul de tratament include calciu, fosfor, vitaminele A, C, E și grupa B în doze specifice vârstei. Se recomandă limitarea sării de masă și includerea în dietă a alimentelor care au efect alcalinizant, precum și a celor bogate în potasiu. În faza de remisie se prescriu masaj și băi de pin sare. Corecția chirurgicală pentru boala De Toni-Debreu-Fanconi este recomandabilă numai atunci când se dezvoltă deformări osoase severe și se obține o remisiune clinică și biochimică stabilă în decurs de 2 ani.
Sindromul Fanconi (diabet zaharat glucoza-fosfat-amina, boala de Toni-Debreu-Fanconi, sindromul Fanconi izolat primar) - boala genetica, dezvoltat ca urmare a unei mutații autosomal recesive, caracterizată prin reabsorbția afectată a apei și a bio. substanțe active din urina primara (reabsorbtie tubulara), datorita afectarii tubilor renali. Aparține unui grup de boli asemănătoare rahitismului în care apar modificări metabolice sistemice.
Motive
Modificările patologice reprezintă una dintre formele de hiperparatiroidism - o tulburare endocrină care se dezvoltă cu producția excesivă de hormon paratiroidian (produs de glandele paratiroide) ca urmare a hiperplaziei glandulare sau a leziunii maligne.
Opțiuni de moștenire pentru sindromul de Toni-Debreu-Fanconi:
- Autosomal dominant - gena defectuoasă este moștenită de la unul dintre părinți (forma familială).
- Autosomal recesiv – gena defectuoasă este prezentă la ambii părinți. În cazul sindromului despre care vorbim forma locala moștenire autosomal recesivă (cromozomul 15q15.3.)
Sindromul Fanconi la copii poate fi o componentă a altor boli genetice:
- Cistinoza este o acumulare excesivă de cistină (un aminoacid) în citoplasmă (mediul celular fluid intern) al celulei.
- - o încălcare a conversiei galactozei (monozaharide) în glucoză, cauzată de o mutație a genei responsabile de producerea galactozei-1-fosfat uridil transferazei (enzimă).
- Tirozinemia de tip I este un deficit de fumarylacetoacetat hidrolaza, care duce la afectarea metabolismului tirozinei.
- - distrofie hepatocerebrala severa cauzata de metabolismul afectat al cuprului.
- fructoză - pierderea enzimei ca urmare a absorbției afectate a fructozei, intoleranța acesteia cauzată de o deficiență a proteinei transportoare de fructoză.
S-a stabilit că patologia se bazează pe tubulopatia combinată - un grup de boli în care transportul substanțelor biologic active în sistemul tubular este afectat. Veriga principală în mecanismul de dezvoltare a sindromului Fanconi este un defect în mitocondriile (depozitul de energie al celulei) în ciclul acidului tricarboxilic (ciclul Krebs), care este o etapă cheie a respirației celulare.
Etapele mecanismului de dezvoltare a bolii, în care fiecare etapă ulterioară va fi o consecință a celei anterioare, pot fi prezentate după cum urmează:
- Defect mitocondrial, tubulopatie enzimatică.
- Reabsorbția afectată a aminoacizilor și a enzimelor în tubii renali.
- Acumulare de acid ().
- Resorbția osoasă (distrugerea).
- Reabsorbție afectată a calciului și potasiului în tubuli.
Celulele pierd aportul de energie, ducând la tulburări metabolice severe. Factorii de risc includ:
- intoxicații cu metale grele;
- infecții toxice;
- luarea de antibiotice tetracicline expirate;
- deficit de vitamina D;
- (tulburarea metabolismului proteic).
Clasificare
Există forme primare (idiopatice) și secundare ale bolii Fanconi. Forma primară se dezvoltă ca urmare a moștenirii unei gene defecte. Forma secundară apare cu alte boli congenitale, determinate genetic.
Sindromul secundar poate apărea pe fondul patologiilor dobândite:
- paraproteinemie - prezența corpurilor proteice anormale în sânge;
- - tulburari severe care se dezvolta atunci cand glomerulii rinichilor sunt afectati;
- tubulointerstițial - un grup de boli de rinichi caracterizate prin afectarea primară a tubilor;
- malignitate(sindrom paraneoplazic);
- în caz de otrăvire;
- arsuri severe.
Simptome
Sindromul Fanconi, ale cărui simptome apar la copii până în al doilea an de viață, are două opțiuni de dezvoltare, în funcție de parametrii clinici și de laborator:
- Prima opțiune este caracterizată printr-o evoluție severă, întârziere severă în dezvoltarea fizică, fracturi și deformări osoase ca urmare a hipocalcemiei și absorbție afectată în intestin.
- A doua opțiune este un curs relativ ușor, semne moderate de întârziere a dezvoltării fizice, deformări osoase cu niveluri normale de calciu, absorbția calciului în intestine este în limite normale.
Primele simptome ale bolii la copiii sub doi ani:
- o scădere bruscă a apetitului;
- deficit de greutate corporală;
- letargie;
- (tulburări digestive cauzate de deficitul proteico-energetic);
- sete;
- tensiune arterială scăzută;
- (cantitate mare de urină);
- febră de grad scăzut;
- vărsături.
Astfel de copii nu pot merge până la vârsta de aproximativ cinci ani.
La apogeul bolii, până la vârsta de cinci sau șase ani, primul semn va fi (înmuierea oaselor), deformările osoase, paralizia asociată cu deficiența de calciu.
După ce apar primele semne, există o întârziere în dezvoltarea mentală și fizică. Decalcificarea generalizată (răspândită în întregul corp) se manifestă prin deformări ale extremităților inferioare ( hallux valgus- curbura spre interior, varus - curbura spre exterior), pierderea tonusului muscular, curbura toracelui, oasele antebratelor si umerilor. Lipsa fosforului la copii duce la apariția.
Pe măsură ce sindromul progresează, copiii dezvoltă tulburări vizuale, boli sistemul nervos, urinare și sistemul digestiv, boli ale organelor ORL. Apar rar.
Cu sindromul Fanconi la adulți, osteomalacia apare din cauza deficitului de minerale și oligoelemente. Pacienții se plâng de dureri osoase, slăbiciune musculară, letargie și posibil crescute tensiunea arterială, dezvoltarea insuficienței renale în absența terapiei.
La copii vârstă fragedă chiar și în primele săptămâni de viață, pot apărea semne ale sindromului Wissler-Fanconi din cauza unor reactie alergica. Patologia se caracterizează prin febră, erupții cutanate eritematoase și leziuni ale articulațiilor (de obicei, mâinile).
Diagnosticare
Pentru depistarea bolii Fanconi se folosesc metode de diagnostic de laborator și imagistic. Cercetare biochimică sângele vă permite să identificați lipsa de calciu și fosfor, beta-2-microglobulină (proteine cu molecul scăzut). Urina dezvăluie aminoacidurie (produse metabolice aminoacizilor), renale (tulburări electrolitice), glicozurie (zahăr în urină), o cantitate mare de fosfați și deficit de microelemente (sodiu, calciu, potasiu, fosfor și altele).
Examenul cu ultrasunete și RMN joacă un rol important în evaluarea funcției renale.
Examinarea cu raze X a oaselor face posibilă studierea structurii osoase, detectarea tulburărilor structurale, gradul de osteoporoză, deformare și evaluarea decalajului legat de vârstă în dezvoltarea țesutului osos. În boala Fanconi, radiografia relevă:
- structura osoasă fibroasă grosieră;
- - distrugerea plăcii de creștere epifizare (cartilaginoasă);
- structura celulară și creșteri asemănătoare pintenilor în tibiei;
- și fracturi – în stadii ulterioare.
Un studiu de emisie de pozitroni (PET) detectează acumularea unei substanțe radioizotopice în zonele de creștere ale oaselor pacientului.
Materialul de biopsie dezvăluie o încălcare a structurii osoase, lacune (depresiuni patologice) și mineralizare slabă.
Pentru diabetul cu glucoză-fosfat-amină, se efectuează diagnostic diferenţial cu următoarele patologii:
- cistinoză;
- glicogenoza;
- diverse sindroame (scăzut, nefrotic);
- mielom multiplu (boală malignă a sângelui);
- intoleranta la fructoza datorita factor genetic, altele boli ereditare;
- afecțiuni apărute în timpul transplantului de rinichi.
Tratament
Sindromul Fanconi este tratat de un hematolog și genetician. În caz de anomalii în structura structurilor renale, proteinurie severă, este necesară consultarea unui medic urolog și nefrolog, în caz de tulburări endocrine - un endocrinolog, în caz de deficiență de vedere - un oftalmolog.
Terapia vizează:
- eliminarea tulburărilor electrolitice, în special a acidozei tubulare;
- corectarea dezechilibrului acido-bazic;
- eliminare manifestări clinice(terapie simptomatică).
Cursul prescrie suplimente de potasiu și calciu, vitamina D cu o creștere treptată a dozei și teste de sânge în timp pentru conținutul de fosfor și calciu. Recomandat bea multe lichide si dieta. În dietă, ar trebui să limitați consumul de alimente sărate și sare, să introduceți lapte, caise uscate, prune uscate, sucuri de fructe. Când numărul de sânge este normalizat, puteți face masaj și face băi de pin.
Intervenția chirurgicală este indicată numai pentru deformările osoase severe. Operația se efectuează cu remisiune stabilă de la un an și jumătate, ceea ce este confirmat de indicatorii de diagnostic și manifestările clinice.
Cel mai mult complicatie severa diabet zaharat glucoză-fosfat-amină - . În cazul unei boli severe care amenință viața pacientului, este indicată hemodializa („rinichi artificial”).

Pentru unele tipuri de insuficiență renală, hemodializa este efectuată temporar până la ameliorare sau recuperare. functia renala. În alte cazuri, cu procese ireversibile în rinichi, procedura se efectuează pe viață.
Dializa implică trecerea sângelui sistem special, unde biofluidele sunt separate substante toxice care sunt îndepărtate cu ajutorul dializatului. Organismul este eliberat de produsele de degradare toxice până la următoarea acumulare.
Prognoze
Prognosticul depinde de boala de bază împotriva căreia s-a dezvoltat sindromul. Dacă boala de bază este un neoplasm, dacă este îndepărtată cu succes, prognosticul poate fi relativ favorabil.
| Sindromul De Toni-Debreu-Fanconi | |
|---|---|
| ICD-10 | 72.0 72.0 |
| ICD-9 | 270.0 270.0 |
| OMIM | , , , , Și |
| BoliDB | |
| MedlinePlus | |
| eMedicine | ped/756 |
| Plasă | D005198 |
Poveste
Tipul de moștenire este autosomal recesiv, a fost identificată și o formă autosomal dominantă cu localizarea genei pe cromozomul 15q15.3. Expresivitatea genei mutante în starea homozigotă variază semnificativ. Există cazuri sporadice cauzate de o nouă mutație. Se crede că boala se bazează pe defecte determinate genetic în fosforilarea enzimatică în tubii renali (tubulopatie combinată), deficit de enzime ale complexelor 2 și 3 ale lanțului respirator - succinat dehidrogenază și citocrom oxidază. Oamenii de știință clasifică boala ca o boală mitocondrială [ ] .
Patogeneza
Modificările patologice reprezintă una dintre variantele hiperparatiroidismului secundar. Veragă principală în patogeneză este un defect al enzimei mitocondriale în ciclul Krebs, o tubulopatie enzimatică caracterizată prin reabsorbția afectată a glucozei, aminoacizilor, fosfaților și bicarbonaților în tubii renali. Pierderea aminoacizilor și a bicarbonatului contribuie la dezvoltarea acidozei metabolice, pe fondul căreia crește resorbția țesutului osos și scade reabsorbția de potasiu și calciu în tubii renali, ceea ce duce la dezvoltarea hipokaliemiei și hipercalciuriei. Pierderea fosforului duce la dezvoltarea rahitismului, iar la copiii mai mari și la adulți - la osteomalacie.
Astfel, un defect al enzimei mitocondriale în ciclul Krebs duce la întreruperea proceselor de alimentare cu energie pentru reabsorbția fosfaților, glucozei și aminoacizilor în tubii renali și excreția crescută a acestora în urină - echilibrul acido-bazic este perturbat și acidoza metabolică și lipsa de fosfați contribuie la distrugerea țesutului osos asemănător cu modificările rahitismului scheletului și osteomalaciei.
Tabloul clinic
Primele semne ale bolii apar în a doua jumătate a vieții - copiii sunt letargici, hipotrofici, apetitul este redus brusc, se observă vărsături, febră scăzută, hipotensiune arterială, sete, poliurie, deshidratare. Un complex de simptome complet dezvoltat se formează în al doilea an de viață. Dacă boala se manifestă la vârsta de 5-6 ani, atunci primele semne sunt simptome de osteomalacie, deformare osoasă și paralizie hipokaliemică. Din cel de-al doilea an de viață, se detectează un întârziere în dezvoltarea fizică și intelectuală, apare decalcifiere generalizată, manifestată prin deformări osoase ale picioarelor (valgus sau varus), toracelui, antebrațelor și humerului, și scăderea tonusului muscular. Razele X dezvăluie deformări ale oaselor, coloanei vertebrale, fracturi, osteoporoză sistemică de severitate diferită, subțierea stratului cortical al oaselor tubulare, slăbirea zonelor de creștere și o întârziere a ratei de creștere a țesutului osos față de vârsta nominală a copilului. Oasele devin fragile.
Examenul de laborator evidențiază normo- sau hipocalcemie, hipofosfatemie, nivel crescut fosfataza alcalina. Ca urmare a scăderii reabsorbției bicarbonaților în tubii renali, se observă acidoză hipercloremică pe fondul excesului de hormon paratiroidian și a normo- sau hipocalcemiei. Analiza biochimică a urinei relevă aminoacidurie, glucozurie (cu niveluri normale glicemie), natriurie, hipocalciurie pe fondul hiperfosfaturiei.
În funcție de severitatea manifestărilor clinice și a tulburărilor metabolice, se disting două variante clinice și biochimice ale bolii:
- Primul caracterizată prin întârziere semnificativă a dezvoltării fizice, boală severă cu deformări osoase severe și adesea fracturi osoase, hipocalcemie severă (1,6-1,8 mmol/l) și scăderea absorbției de calciu în intestin.
- La doileaÎn această variantă se constată o întârziere moderată a dezvoltării fizice, o evoluție ușoară cu deformări osoase minore, normocalcemie și absorbție normală a calciului în intestin.
Tulburări biochimice
- scăderea nivelului de calciu în sânge;
- scăderea nivelului de fosfor din sânge;
- niveluri crescute ale fosfatazei alcaline;
- dezvoltarea acidozei metabolice (pH: 7,35...7,25; BE: −10...-12 mmol/l) ca urmare a unui defect în reabsorbția bicarbonaților în tubii proximali;
- excreția urinară normală de calciu;
- clearance-ul crescut al fosfaților urinari, absorbția fosfaților în intestin nu este afectată;
- dezvoltarea glucozuriei (20-30 g/l și mai sus);
- dezvoltarea hiperaminoaciduriei generalizate;
- disfuncție a acidogenezei amoniacului - scăderea acidității de titrare, creșterea pH-ului urinei peste 6,0;
- dezvoltarea hipokaliemiei.
Rezultatul bolii este dezvoltarea insuficienței renale cronice.
Diagnostic diferenţial
Diagnosticul diferențial al sindromului de Toni-Debreu-Fanconi se realizează cu