Deficitul selectiv de IgA este cea mai frecventă afecțiune de imunodeficiență primară (PIDS). Incidența pacienților cu deficit selectiv de IgA variază de la 1:400 la 1:1000 la populația caucaziană și este semnificativ mai mică, de la 1:4000 la 1:20000, la populația mongoloidă. În Statele Unite, prevalența bolii variază de la 1 la 223-1000 în grupul de studiu la 1 la 400-3000 la donatorii de sânge sănătoși. Studii similare nu au fost efectuate în Rusia.
Această afecțiune se caracterizează printr-o scădere selectivă a concentrației serice de IgA sub 0,05 g/l (la copiii cu vârsta peste patru ani) cu nivel normal alte imunoglobuline serice, reacție normală anticorpii serici și răspunsul imun normal mediat de celule. În majoritatea studiilor, frecvența de apariție în rândul bărbaților și al femeilor a fost aproximativ aceeași.
Persoanele cu incapacitatea de a produce IgA pot rămâne asimptomatice din cauza mecanismelor de compensare sau suferă frecvent de afecțiuni respiratorii, digestive sau sistemul genito-urinar, patologie gastroenterologică (de exemplu, boala celiacă), o tendință la tulburări atopice, cum ar fi febra fânului, astm bronsic, dermatită atopică, mediată de IgE alergie la mancare, precum și boli neurologice și autoimune (cel mai adesea acestea sunt artrita reumatoida, lupus eritematos sistemic, purpură trombocitopenică idiopatică, sindrom Sjogren). Pentru deficitul selectiv de IgA boli alergice, precum dermatita atopică și astmul bronșic, au apărut în 40% din cazuri (Consilium Medicum, 2006). De asemenea, tipice pentru majoritatea acestor pacienți sunt reacțiile anafilactice în timpul transfuziei componentelor sanguine și administrarea de imunoglobuline intravenoase, care este asociată cu prezența IgA în aceste produse.
Simptomele clinice ale deficitului selectiv de IgA pot apărea în copilăria timpurie, dar odată cu vârsta, frecvența și severitatea infecțiilor transmise pot scădea datorită creșterii compensatorii a anticorpilor din subclasele IgG1 și G3, IgM. O altă explicație pentru absență simptome clinice Poate exista un nivel normal al IgA secretoare, în ciuda unei scăderi a nivelurilor de imunoglobuline serice. Sau, dimpotrivă, unii pacienți cu deficit de IgA selectiv diagnosticat inițial pot dezvolta un tablou clinic al deficienței imune variabile comune.
Terapia pentru deficitul selectiv de IgA constă în prezent în identificarea boli concomitente, efectuând măsuri preventive acceptare pentru a reduce riscul de infecție, precum și pentru a trata rapid și eficient infecțiile.
Nu există un tratament specific. Prognosticul pentru pacienții cu deficit de IgA este în general bun, cu excepția cazului în care sunt semnificative manifestari clinice. Deficitul de IgA la copii poate fi corectat în timp.
Fiind determinate genetic, stările de imunodeficiență apar din cauza defectelor aparatului genetic. Pacienții cu deficiență imună variabilă comună și cei cu deficit selectiv de IgA se găsesc adesea în aceeași familie și au un haplotip HLA comun; multe au alele rare și deleții de gene din clasa MSI 3 pe cromozomul 6. Recent, unele cazuri familiale de deficiență imună variabilă comună și deficiență selectivă de IgA s-a dovedit a fi cauzate de o mutație a genei TNFRSF13B, care codifică o proteină cunoscută sub numele de TACI (activator transmembranar și modulator de calciu și interactor ciclofilină-ligand). Este probabil ca în cazurile în care nu a fost găsită nicio mutație TACI, boala ar putea fi cauzată de mutații spontane sau ereditare ale altor gene care nu au fost încă documentate.
În prezent, posibilele manifestări clinice ale deficitului selectiv de IgA, variantele de curs și posibilele boli concomitente au fost descrise suficient de detaliat. Hotărâtoare în diagnosticul bolii este o scădere selectivă a concentrației serice de IgA la copiii începând cu vârsta de 4 ani sub 0,05 g/l cu niveluri normale ale altor imunoglobuline serice în imunograme repetate. Tratamentul constă în identificarea bolilor concomitente, luarea de măsuri preventive pentru reducerea riscului de infecție și necesită, de asemenea, prompt și tratament eficient boli infecțioase.
Nu există informații despre frecvența de apariție a acestei stări primare de imunodeficiență în populația rusă, ceea ce nu face posibilă compararea prevalenței bolii în țara noastră cu alte țări în care au fost deja efectuate studii similare.
Principala problemă este lipsa unor recomandări uniforme pentru managementul pacienților cu deficit selectiv de IgA.
Pentru a evalua incidența deficitului selectiv de IgA în rândul copiilor din grup observarea dispensarului„copii frecvent bolnavi” și caracterizează gama manifestărilor sale clinice în Federația Rusă pe baza instituției bugetare federale de stat „FNKTs DGOI numită după Dmitri Rogachev” a Ministerului Sănătății al Federației Ruse și a instituției bugetare de stat a Spitalului Clinic Orășenesc pentru Copii nr. 9 numită după. Această lucrare a fost realizată de Departamentul de Sănătate G.N. Speransky.
Materiale și metode de cercetare
Obiectul studiului l-au constituit copiii cu deficit selectiv de IgA, observați în Spitalul Clinic Orășenesc de Copii Nr. 9 care poartă numele. G. N. Speransky DZM. În plus, a fost efectuată o analiză retrospectivă documentatie medicala pentru perioada 2003-2010. 9154 de pacienți din grupa de observare a dispensarului „copii bolnavi frecvent” (Tabel 1-3).
În timpul examinării au fost utilizate următoarele metode:
- clinic și anamnestic;
- generală şi teste biochimice sânge;
- studiul imunologic al compoziției sângelui folosind metode de nefelometrie și citometrie în flux;
- teste de scarificare;
- determinarea IgE specifice prin imunoblot;
- studiul funcției respirației externe;
- studiu rinocitologic.
Diagnosticul deficitului selectiv de IgA a fost pus pe baza unei scăderi selective a concentrației serice de IgA sub 0,05 g/l cu indicatori normali alte imunoglobuline serice în imunograme repetate și excluzând altele motive posibile deficienţa lor la copiii cu vârsta peste 4 ani.
La colectarea anamnezei Atentie speciala concentrat pe frecvența și gama manifestărilor clinice, patologie concomitentă, iar istoria familiei a fost, de asemenea, studiată în detaliu. Examinarea clinică a copiilor a fost efectuată în conformitate cu metodele general acceptate. Conținutul de imunoglobuline din clasele A, G, M, E în ser a fost determinat prin nefelometrie pe un nefelometru BN 100 (Dade Bering, Germania) utilizând un kit Dade Behring. Fenotiparea limfocitelor a fost efectuată prin citometrie în flux pe un dispozitiv FacsScan (Becton Dickenson, SUA) utilizând anticorpi monoclonali marcați fluorescent Simultest (Becton Dickenson, SUA). Pacienții cu orice manifestări de atopie, precum și toți pacienții cu nivel crescut IgE, care a fost identificat ca urmare a evaluării indicatorilor starea imunitară prin metoda nefelometriei s-a efectuat un examen alergologic suplimentar folosind metoda testelor de scarificare la copii cu vârsta peste 4 ani sau prin metoda determinării IgE specifice în serul sanguin al pacienților sub 4 ani. Copiii cu diagnostic de astm bronșic sau antecedente de sindrom bronho-obstructiv au fost supuși unui studiu al funcției respiratorii externe folosind aparatul Spirovit SP-1 (Schiller AG, Elveția). De asemenea, au fost efectuate toate examinările și consultările suplimentare necesare cu specialiștii înrudiți, ținând cont de reclamațiile existente.
Rezultatele și discuția lor
O analiză retrospectivă a fișelor medicale ale pacienților cu diagnostic de referință de „ARVI recurent”, „CHD”, „CHD”, precum și „EDD” a permis să se stabilească că frecvența deficitului selectiv de IgA la acest grup de copii este de două sau chiar de trei ori mai mare decât în populaţie.
Numărul absolut, precum și procentul de copii cu această imunodeficiență primară pe an, pot fi văzute în tabel. 4.
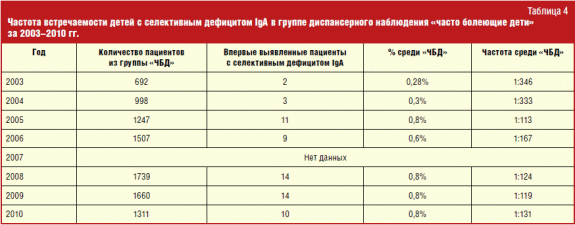
Din păcate, datele pentru 2007 nu sunt disponibile. În 2003 și 2004 Au fost consultați 692 și 998 de copii. Dintre aceștia, au fost identificați un total de 5 pacienți cu deficit selectiv de IgA, ceea ce este puțin mai frecvent decât media populației - 1:346 și, respectiv, 1:333, față de 1:400-600. Din 2005, frecvența pacienților nou diagnosticați cu această BIP a crescut brusc: 1:113 în 2005, 1:167 în 2006, 1:124 în 2008, 1:119 în 2009 și, în final, 1:131 în 2010. în studiu, frecvența de apariție s-a modificat de la 1:346 în 2003 la 1:131 în 2010, când a fost cea mai mare în comparație cu anii precedenți. Creșterea incidenței pacienților cu deficit selectiv de IgA în al treilea an de la începerea activității ar trebui asociată cu vigilența crescută a medicilor cu privire la această patologie, precum și cu îmbunătățirea diagnostic de laborator. Este necesar să se extindă în continuare cunoștințele medicilor despre această boală, deoarece fluxul de copii ai căror părinți aduc la imunolog plângeri de boli frecvente, crește de la an la an.
În timpul acestei lucrări, au fost examinați prospectiv 235 de copii și 32 de adulți.
Grupul principal a fost format din 73 de copii diagnosticați cu deficit selectiv de IgA.
Al doilea grup de pacienți a inclus 153 de copii cu purpură trombocitopenică idiopatică (ITP). S-a efectuat o evaluare a stării imunitare a pacienților cu ITP pentru a identifica deficitul selectiv de IgA în rândul acestora, deoarece această corelație este descrisă în literatura mondială și aceleași date au fost obținute în timpul acest studiu. Nu am identificat un singur copil cu absența IgA printre ei. În ciuda faptului că la examinarea stării imune a pacienților cu ITP, nu am putut identifica deficitul selectiv de IgA printre aceștia, au fost identificate și alte defecte umorale minore: deficiența subclaselor de IgG, hipogamaglobulinemie infantilă, scăderea parțială a IgA.
Al treilea grup a inclus 32 de adulți cu vârsta cuprinsă între 20 și 54 de ani, precum și 8 copii cu vârsta cuprinsă între 4 și 10 ani, care erau rude apropiate ale pacienților cu deficit selectiv de IgA, a căror stare imunitară a fost evaluată pentru a căuta și a descrie cazurile familiale.
În cadrul anchetei și analizei datelor obținute s-au obținut rezultatele descrise mai jos.
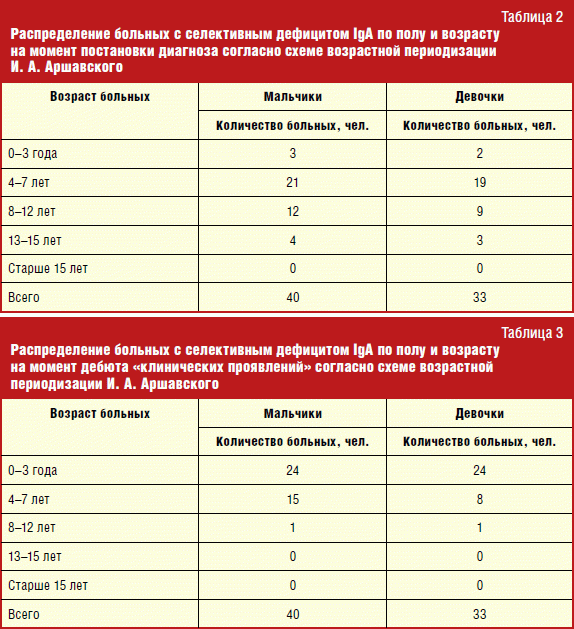
Raportul dintre bărbați și femei în rândul pacienților cu deficit selectiv de IgA a fost aproximativ același. Au fost examinați 40 de băieți și 33 de fete. Aceasta corespunde datelor literaturii mondiale.
Detectarea de vârf a deficitului selectiv de IgA a avut loc la vârsta de 4-7 ani. Bolile infecțioase repetate au apărut de obicei în vârstă fragedă sau odată cu începerea frecventării unei instituții preșcolare. De regulă, înainte de a merge la imunolog, copiii au acumulat un anumit istoric infecțios, deoarece există anumite semne care fac posibilă bănuiala că au BIP. Și, în plus, chiar dacă studiul a fost efectuat la o vârstă mai fragedă și a evidențiat absența IgA până la vârsta de 4 ani, acest lucru nu ne-a permis să facem un diagnostic clar de PIDS; nu am putut exclude complet imaturitatea sistemul de sinteză a imunoglobulinelor. Prin urmare, până la vârsta de 4 ani, diagnosticul a fost pus pe bază de întrebări și s-a recomandat observația dinamică. Prin urmare, intervalul este de 4-7 ani, respectiv.
Principalele plângeri la tratarea copiilor cu deficit selectiv de IgA au fost cele respiratorii frecvente infecții virale cu un curs necomplicat. Debutul bolilor respiratorii recurente, de regulă, a avut loc înainte de vârsta de 3 ani. Aceasta corespunde și cu datele literaturii mondiale. Deoarece monitorizarea dinamică a majorității pacienților din studiul nostru a fost efectuată pentru o perioadă lungă de timp, timp de câțiva ani, uneori înainte ca pacientul să treacă la vârsta adultă, se poate susține că odată cu vârsta, frecvența și severitatea infecțiilor au scăzut. Probabil că acest lucru s-a întâmplat din cauza unei creșteri compensatorii a anticorpilor din subclasele IgG1 și IgG3, IgM, dar această problemă necesită studii suplimentare. A doua cea mai frecventă plângere la tratament au fost infecțiile virale respiratorii acute frecvente, însoțite de complicații. Frecvența infecțiilor virale respiratorii acute complicate, atipice, odată cu vârsta, la pacienții noștri, după cum arată observația dinamică, a scăzut și ea.
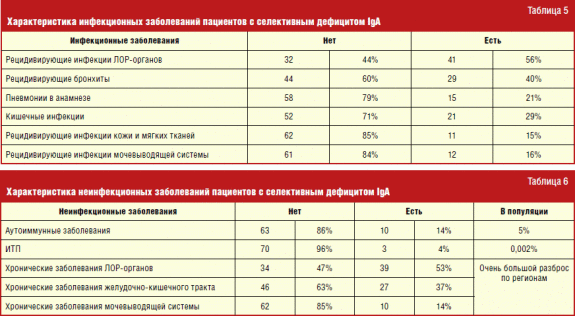
Dintre spectrul bolilor infecțioase la pacienții cu deficit selectiv de IgA, locul principal a fost ocupat de bolile infecțioase ale organelor ORL și infecțiile inferioare. tractului respirator. Acest lucru se datorează faptului că o scădere a IgA secretoare, care face parte din imunitatea locală, duce la o infecție ușoară și la proliferarea microorganismelor pe membranele mucoase, care sunt cele mai vulnerabile atunci când sunt în contact cu boli infecțioase transmis prin picături în aer.
Pe spectru boli necontagioase S-a identificat o corelație evidentă cu bolile autoimune, care sunt cele mai importante manifestări ale deficitului selectiv de IgA, în special cu purpura trombocitopenică idiopatică (1,5-2 la 100 mii).
Din boală autoimună la pacienții cu deficit selectiv de IgA, cele mai frecvente tipuri au fost artrita reumatoidă juvenilă (de 4 ori), purpura trombocitopenică cronică idiopatică (de 3 ori) și hepatita autoimună (de 3 ori). În plus, conform literaturii mondiale, pacienții cu deficit selectiv de IgA au o frecvență crescută a afecțiunilor autoimune în rândul familiei lor apropiate. Dar, conform cercetării noastre, numărul acestora nu a depășit valorile populației generale.
Frecvența bolilor atopice în rândul pacienților cu deficit selectiv de IgA a fost semnificativ mai mare decât în populație (Tabelul 4). Numai frecventa rinită alergică comparabil cu populația generală. Observații similare sunt reflectate într-o serie de studii anterioare. Nu se poate spune că bolile alergice la majoritatea pacienților cu deficit de IgA sunt mai severe decât la persoanele fără acest defect imunologic. Cu toate acestea, prevalența ridicată a atopiei dă naștere la întrebarea efectuării unui examen imunologic în vederea identificării formelor de deficit selectiv de IgA, care nu s-au manifestat încă clinic. Deși acest lucru poate să nu aibă un rol decisiv în abordarea terapiei pentru starea atopică actuală, va ajuta la stabilirea unui diagnostic în timp util și la reducerea riscurilor posibile pentru persoanele care au deficiență selectivă de IgA.
La analizarea imunogramelor repetate în timpul observării dinamice la copiii cu deficit selectiv de IgA, din cauza modificărilor persistente parametrii de laborator, doi au fost identificati grupuri mari pacientii. În grupul A, a existat o absență a IgA fără alte modificări. În grupul B, absența IgA a fost combinată cu o creștere persistentă a nivelului de IgG. A fost efectuată o analiză comparativă a acestor grupuri de pacienți.

Vârsta de debut a manifestărilor clinice la aceste grupuri nu a diferit semnificativ.
S-a constatat că la pacienții cu deficit selectiv de IgA, o creștere a nivelului de IgG se corelează cu boli infecțioase recurente ale pielii și țesuturilor moi. Această problemă necesită studii suplimentare.
La compararea acestor grupuri de pacienți, nu au fost identificate diferențe semnificative în spectrul alergopatologiei.
În timpul lucrărilor, starea imună a fost evaluată la 20 de familii de pacienți cu deficit selectiv de IgA. Au fost identificate patru cazuri familiale. În plus, a fost colectat un istoric familial detaliat. Printre rudele adulte cu antecedente infecțioase împovărate care au putut fi supuse examinării, au apărut anumite tulburări imunitate umorală. În consecință, atunci când sunt detectate defecte umorale minore (în special, deficit selectiv de IgA), examinarea rudelor apropiate, în special în prezența unui istoric infecțios împovărat, este obligatorie.
Datorită faptului că deficiența selectivă de IgA în rândul copiilor din grupul de urmărire „copii frecvent bolnavi” este mult mai frecventă decât în populația pediatrică generală, pediatrii practicieni trebuie să fie atenți la această boală. Nu este întotdeauna ușor de bănuit, deoarece manifestările clinice sunt foarte variabile: de la forme asimptomatice la cele recurente. infecții bacteriene cu nevoie de frecvente terapie antibacteriană. Se recomandă extinderea cunoștințelor medicilor pediatri și specialiștilor din ambulatoriu și internați despre defecte minore ale sistemului imunitar umoral.
Deoarece printre pacienții cu deficit selectiv de IgA frecvența patologiei alergice (astm bronșic, dermatită atopică, alergie alimentară) este semnificativ mai mare, frecvența bolilor autoimune și a bolilor hematologice este mai mare, precum și frecvența. boli cronice(organe ORL, sistemul genito-urinar, tract gastrointestinal) decât la populație, identificarea acestuia este obligatorie pentru a oferi cuprinzătoare și în timp util îngrijire medicală pacientii.
Se recomandă trimiterea copiilor cu antecedente infecțioase împovărate, a pacienților cu boli hematologice și autoimune pentru consultarea unui imunolog/examen imunologic și efectuarea unui examen de screening a nivelului de IgA totală la pacienții cu boli alergice.
Studiul a constatat că la majoritatea copiilor cu deficit selectiv de IgA a existat o corelație între prezență patologie autoimunăși o creștere persistentă a IgG în imunogramele repetate. Nu a fost stabilită o astfel de corelație pentru alte boli. Astfel de modificări ale indicatorilor sunt un factor de risc pentru dezvoltarea patologiei autoimune la un copil și necesită o atenție specială.
În ciuda faptului că nu a fost stabilită o corelație între prezența unui istoric familial de deficit selectiv de IgA și severitatea manifestărilor clinice la pacienți, pentru acești pacienți, examinarea rudelor apropiate, mai ales în prezența unui istoric infecțios împovărat, este obligatoriu.
Literatură
- Hammarstrom L., Lonnqvist B., Ringden O., Smith C. I., Wiebe T. Transferul deficitului de IgA la un pacient grefat de măduvă osoasă cu anemie aplastică // Lancet. 1985; 1 (8432): 778-781.
- Latiff A. H., Kerr M. A. Semnificația clinică a deficitului de imunoglobuline A // Annals of Clinical Biochemistry. 2007; 44 (Pt 2): 131-139.
- Al-Attas R. A., Rahi A. H. Deficitul primar de anticorpi la arabi: primul raport din estul Arabiei Saudite // Journal of Clinical Immunology. 1998; 18 (5): 368-371.
- Carneiro-Sampaio M. M., Carbonare S. B., Rozentraub R. B., de Araujo M. N., Riberiro M. A., Porto M. H. Frecvența deficitului selectiv de IgA în rândul donatorilor de sânge brazilieni și al femeilor însărcinate sănătoase // Alergologie Imunopatologie (Madr). 1989; 17 (4): 213-216.
- Ezeoke A.C. Deficiența selectivă de IgA (SIgAD) în Nigeria de Est // Jurnalul African de Medicină și Științe Medicale. 1988; 17 (1): 17-21.
- Feng L. Studiu epidemiologic al deficitului selectiv de IgA la 6 naționalități din China // Zhonghua Yi Xue Za Zhi. 1992; 72 (2): 88-90, 128.
- Pereira L. F., Sapina A. M., Arroyo J., Vinuelas J., Bardaji R. M., Prieto L. Prevalența deficitului selectiv de IgA în Spania: mai mult decât am crezut // Sânge. 1997; 90(2):893.
- Wiebe V., Helal A., Lefranc M. P., Lefranc G. Analiza moleculară a deleției multigene ale imunoglobulinei CH T17 (del A1-GP-G2-G4-E) // Human Genetics. 1994; 93(5):5.
L. A. Fedorova*,
E. S. Pushkova*
I. A. Korsunsky**, 1,candidat Stiinte Medicale
A. P. Prodeus*,Doctor în Științe Medicale, Profesor
Imunodeficiență - o scădere a indicatorilor cantitativi și/sau a activității funcționale a principalelor componente sistem imunitar, ceea ce duce la perturbarea apărării organismului împotriva microorganismelor patogene și se manifestă prin creșterea morbidității infecțioase.
După cum se știe, funcția principală a sistemului imunitar este recunoașterea și eliminarea substanțelor străine de natură antigenică care pătrund în organism din mediu inconjurator(microorganisme) sau care apar pe cale endogenă (celule tumorale). Această funcție este implementată folosind factori imunitatea înnăscută(fagocitoză, peptide antimicrobiene, proteine din sistemul complementului, sistemul de celule NK etc.) și imunitatea dobândită sau adaptativă, realizată cu ajutorul răspunsurilor imune celulare și umorale. Reglarea activității componentelor de apărare imună a organismului și interacțiunea lor are loc cu ajutorul citokinelor și a contactelor intercelulare.
În fiecare dintre componentele enumerate ale sistemului imunitar, precum și în mecanismele de reglare a acestora, pot apărea tulburări, ducând la dezvoltarea imunodeficienței, a cărei manifestare clinică principală este sensibilitate crescută la agenții patogeni ai bolilor infecțioase. Există 2 tipuri de imunodeficiențe: primare și secundare.
Imunodeficiențe primare(PID) - boli ereditare cauzate de defecte ale genelor care controlează răspunsul imun. BIP-urile sunt boli care variază ca natură și severitate a defectelor imune, a manifestărilor clinice și a anomaliilor moleculare. Tabloul clinic al BIP se caracterizează prin repetate și cronice, severe procese infecțioase, în principal sistemul bronhopulmonar
și organe ORL, piele și mucoase; Se pot dezvolta limfadenită purulentă, abcese, osteomielita, meningită și sepsis. În unele forme există manifestări de alergii, boli autoimune și posibila dezvoltare a unora tumori maligne. Ar trebui să acordați atenție decalajului indicatori de vârstă dezvoltarea fizică. În prezent, au fost descrise aproximativ 80 de BIP și au fost identificate genele responsabile pentru dezvoltarea majorității acestor boli. Testele de laborator adecvate fac posibilă diferențierea patologiei la nivelul limfocitelor și a patologiei la nivelul mecanismelor nelimfocitare de distrugere și îndepărtare a antigenelor.
Prevalența PID depinde de forma bolii și în medie variază de la 1:10.000 la 1:100.000 de nou-născuți. Deficiență selectivă IgA, de exemplu, este mult mai frecventă, variind de la 1:500 la 1:1500 de persoane în populația generală. Prevalența diferite forme PID variază în tari diferite. Cele mai frecvente defecte în formarea anticorpilor sunt 50-60% din cazuri, PID combinat - 10-30%, defecte de fagocitoză - 10-20%, defectele complementului - 1-6%. Majoritatea BIP se manifestă în copilăria timpurie, deși debutul mai târziu este posibil pentru unele forme de BIP, în special pentru deficiența imunologică variabilă comună (CVID).
Conform mecanismelor de dezvoltare, există 4 grupuri principale de PID:
Primul grup - predominant umoral sau cu celule B
PID;
Grupa 2 - PID combinat (toate imunodeficiențele celulelor T au funcția afectată a celulelor B);
Grupa 3 - PID cauzată de defecte în fagocitoză;
Grupa 4 - PID cauzată de defecte ale sistemului complement.
Principiile diagnosticului imunodeficiențelor primare
Diagnosticul precoce și inițierea la timp a tratamentului determină prognosticul bolii. Efectuarea unui diagnostic la nivelul pediatrilor locali prezintă anumite dificultăți, care se datorează adesea incapacității de a consulta pacientul în timp util cu un imunolog și de a efectua un examen imunologic special de laborator (Tabelul 11-1). Deși cunoașterea caracteristicilor tabloului clinic al PID și modificări
cercetare în general clinică analize de laborator permiteți să suspectați BIP și trimiteți pacientul către specialiști. Societatea Europeană pentru Imunodeficiență a dezvoltat protocoale pentru diagnosticarea precoce a BIP și a creat, de asemenea baza de date electronica date din Registrul European PID. Algoritmul de diagnosticare PID este prezentat în Fig. 11-1.
Tabelul 11-1. Etape ale examenului imunologic pentru suspiciunea de imunodeficiență
Orez. 11-1. Algoritm pentru diagnosticarea imunodeficiențelor primare
Caracteristici generale ale tabloului clinic al imunodeficiențelor primare
Conducă în tablou clinic PID este așa-numitul sindrom infectios- susceptibilitate crescută la agenții patogeni ai bolilor infecțioase în general, recidivante (recurente) neobișnuit de severe; curs clinic, prezența agenților patogeni atipici (deseori oportuniști) în etiologia bolii. Tipul de agent patogen este determinat de natura defectului imunitar. În cazul unor defecte în formarea anticorpilor, este posibilă identificarea rezistenței la medicamente antibacteriene flora - stafilococi, streptococi, hemophilus influenzae. În deficiența imunitară a celulelor T, pe lângă bacterii, viruși (de exemplu, familia herpesvirusului), sunt detectate ciuperci (Candida spp., Aspergillus etc.), și cu defecte fagocitare - stafilococi, bacterii gram-negative, ciuperci etc.
Cercetare de laborator
Dacă datele clinice sugerează PID, trebuie efectuate următoarele studii:
Determinarea unei hemograme detaliate (indicatorii cantitativi și procentuali ai limfocitelor sunt deosebit de importanți);
Determinarea nivelurilor de IgG, IgA și IgM în serul sanguin;
Numărarea subpopulațiilor de limfocite T și B;
◊ analiză stare functionala fagocite (cea mai simplă și mai informativă analiză este testul de reducere cu albastru de tetrazoliu);
◊ analiza pentru conținutul componentelor principale ale complementului (începând cu C3 și C4);
◊ test pentru infecția HIV (dacă există posibili factori de risc);
◊ studii genetice moleculare când este indicat.
Principii de tratament al imunodeficiențelor primare
Scopul principal al terapiei PID este tratamentul complicațiilor bolii și prevenirea acestora. Această abordare se datorează faptului că defectele sistemului imunitar în PID sunt determinate la nivel genetic. În prezent, se efectuează cercetări intensive asupra genei
o nouă terapie pentru imunodeficiențe, care poate duce la apariția unor metode mai radicale de tratare a acestora.
În funcție de forma BIP, tratamentul constă în terapie de substituție, tratamentul și prevenirea manifestărilor infecțioase, autoimune ale bolii, tratament neoplasme maligneși aplicare metode speciale, inclusiv transplantul de celule stem hematopoietice (în funcție de tipul de PID).
DEFECTE DE IMUNOGLOBULINĂ
Hipogamaglobulinemie tranzitorie la copii
Hipogamaglobulinemia tranzitorie la copii este asociată cu caracteristică fiziologică formarea treptată a sistemului de imunoglobuline. Maturarea formării anticorpilor IgM și IgA este „întârziată” în cea mai mare măsură. La copiii sănătoși, conținutul de IgG maternă scade treptat și după șase luni crește producția propriilor anticorpi IgG. La unii copii, cu toate acestea, creșterea nivelului de imunoglobuline este întârziată. Astfel de copii pot suferi de infecții bacteriene recurente. În aceste cazuri, nu trebuie să apelați la perfuzii de preparate de imunoglobuline donatoare (administrare intravenoasă de imunoglobuline).
Deficit selectiv de imunoglobulina A
Deficiența selectivă a imunoglobulinei A (SD IgA - Deficitul selectiv de IgA) se dezvoltă ca urmare a unui defect genetic tnfrsf13b
sau p). Deficiența de IgA în prezența imunoglobulinelor din alte clase este cea mai frecventă imunodeficiență detectată în populația generală cu o frecvență de 1:500-1500 de persoane (și mai des la pacienții cu alergii). Există deficit selectiv de IgA, adică constând într-o deficiență a uneia dintre subclase (30% din cazuri), și completă (70% din cazuri). Deficiența subclasei IgA2 duce la un tablou clinic mai sever decât deficiența subclasei IgA1. Sunt posibile și combinații ale deficitului de IgA cu alte tulburări: cu un defect în biosinteza IgG și cu anomalii ale limfocitelor T. Marea majoritate a oamenilor cu selectiv
Deficitul de IgA este practic sănătos. Pentru copiii sub 2 ani, deficitul de IgA este o afecțiune fiziologică.
Detectează o scădere a concentrației serice de IgA la<5 мг/дл у детей старше 4 лет; IgG и IgM в норме, количество и соотношение субпопуляций лимфоцитов и их функциональная активность могут быть в норме.
Tabloul clinic. Cu deficit de IgA se pot dezvolta 3 grupe de sindroame patologice: infecțioase, autoimune și alergice. Pacienții cu deficit de IgA sunt predispuși la infecții recurente ale tractului respirator superior și ale sistemului digestiv. Cele mai frecvente și severe sunt diverse boli autoimune (artrita reumatoidă, spondilita anchilozantă, sindromul Sjögren, vasculita cu afectarea vaselor cerebrale, tiroidita autoimună, LES, glomerulonefrita, anemie hemolitică, diabet zaharat tip I, vitiligo etc.). Incidența bolii celiace este de 10 ori mai mare decât la copiii cu IgA normale. Manifestările alergice cel mai frecvent identificate sunt: intoleranța la proteinele din laptele de vacă, dermatita atopică (DA), astmul bronșic.
Tratament. Cazurile asimptomatice nu necesită tratament special; în prezența manifestărilor clinice ale bolilor infecțioase, autoimune și alergice, tratamentul se efectuează în conformitate cu standardele.
Terapia de substituție cu imunoglobuline de la donator nu este indicată nici pentru deficiența selectivă sau completă de IgA, deoarece există o probabilitate mare ca primitorul să dezvolte anticorpi anti-izotipici la IgA și să dezvolte complicații de transfuzie cauzate de aceștia.
Agamaglobulinemie cu deficit de celule B
Agammaglobulinemie legată de X (boala lui Bruton) reprezintă 90% din toate cazurile de agammaglobulinemie. Sunt afectați băieții și fiii (אּ, ρ) purtători ai genei defecte btk (Xq21.3-q22), care codifică proteina tirozin kinaza specifică limfocitelor B Btk (tirozin kinaza lui Bruton- tirozin kinaza lui Bruton). Ca urmare a defectului, căile de semnalizare intracelulară sunt întrerupte, recombinarea lanțurilor grele de imunoglobuline și
transferul celulelor pre-B în limfocitele B. La 10% dintre pacienți, agammaglobulinemia cu deficit de celule B este moștenită autosomal recesiv. În prezent, au fost descrise 6 defecte genetice, inclusiv moleculele receptorului celulelor pre-B, proteina adaptoare a celulelor B citoplasmatice (BLNK) și gena Repetă bogată în leucină care conține 8 (LRRC8).
Date de laborator. Nu există limfocite B periferice. Măduva osoasă conține celule pre-B cu lanțul μ în citoplasmă. Numărările celulelor T și testele funcționale ale celulelor T pot fi normale. IgM și IgA nu pot fi detectate în sânge; IgG poate fi prezentă, dar în cantități mici (0,4-1,0 g/l). Nu există anticorpi la antigenele grupelor de sânge și antigenele de vaccin (tetanos, toxine difterice etc.). Se poate dezvolta neutropenie. Examinarea histologică a țesutului limfoid: nu există centre germinale (germinale) și celule plasmatice în foliculii limfoizi.
Tabloul clinic. Dacă antecedentele familiale sunt necunoscute, diagnosticul devine evident la vârsta medie de 3,5 ani. Boala se caracterizează prin hipoplazie a țesutului limfoid, infecții purulente severe, boli infecțioase ale tractului respirator superior (sinuzită, otită) și inferioară (bronșită, pneumonie); posibil gastroenterită, piodermie, artrită septică (bacteriană sau chlamydia), septicemie, meningită, encefalită, osteomielita. Cei mai frecventi agenți patogeni care provoacă boli ale tractului respirator sunt Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus, diaree bacterii intestinale sau Giardia Giardia lamblia. De asemenea, pacienții cu agammaglobulinemie sunt susceptibili la boli infecțioase cauzate de micoplasme și ureaplasme, care provoacă dezvoltarea pneumoniei cronice, artritei purulente, cistita și abcese ale țesutului subcutanat. Virușii tipici sunt virusurile neurotropice ECHO-19 și coxsackie, care provoacă atât encefalită acută și cronică severă, cât și encefalomielita. Manifestările infecțiilor cu enterovirus pot include sindrom asemănător dermatomiozitei, ataxie, dureri de cap și tulburări de comportament. La copiii bolnavi, atunci când sunt imunizați cu vaccinul poliomielitei viu, de regulă, este detectată eliberarea prelungită a virusului poliomielitei prin membranele mucoase, cu virulența restabilită și în creștere (adică în colectivele de copii).
există un pericol real ca copiii sănătoși să se infecteze cu poliomielita ca urmare a contactului cu un copil imunodeficient vaccinat). Tulburările autoimune în agammaglobulinemie pot fi reprezentate de poliartrita reumatoidă, sindromul asemănător sclerodermiei, scleredemul, colita ulceroasă, diabetul zaharat tip I (datorită predominării răspunsului imun Th1).
Examinare fizică. Atenție la întârzierea dezvoltării fizice, la forma degetelor (degete sub formă de tobe), modificări ale formei toracelui, caracteristice bolilor tractului respirator inferior, hipoplazie a ganglionilor limfatici și amigdalelor.
Tratament.
Terapia de substituție: preparatele de imunoglobuline intravenoase se administrează la fiecare 3-4 săptămâni pe viață. Dozele de imunoglobuline sunt selectate astfel încât să creeze o concentrație în serul pacientului care depășește limita inferioară a normei de vârstă.
Discutarea posibilității terapiei genice - genă Btk clonat, dar supraexpresia sa este asociată cu transformarea malignă a țesutului hematopoietic.
În cazul neutropeniei persistente se folosesc factori de creștere. Dacă apar semne de patologie autoimună, se pot prescrie medicamente cu anticorpi monoclonali (infliximab etc.).
Deficiență imunitară variabilă comună
Deficiența imună variabilă comună (CVID) este un grup de sindroame caracterizate printr-un defect în sinteza anticorpilor și a imunității celulare. Un criteriu de diagnostic fiabil pentru CVID este o scădere semnificativă a conținutului de imunoglobuline a două sau trei izotipuri principale la persoanele de ambele sexe în combinație cu unul dintre următoarele simptome:
Debutul bolii este peste 2 ani;
Lipsa izohemaglutininelor și/sau răspuns scăzut la vaccinare;
Excluzând alte cauze ale agammaglobulinemiei.
La unii pacienți, cauza dezvoltării CVID este mutațiile în genele care codifică moleculele implicate în procesele de maturare și supraviețuire a celulelor B: BAFF-R (Receptorul factorului de activare al celulelor B), Blimp-1 (proteina de maturare indusă de limfocitele B-1)și ICOS (Costimulator inductibil). Există o întrerupere a capacității limfocitelor B de a se diferenția în celule plasmatice, se dezvoltă defecte în formarea anticorpilor, este posibilă disfuncția limfocitelor T și se observă o susceptibilitate crescută la boli infecțioase. Sindromul poate apărea în copilărie timpurie, adolescență sau vârsta adultă tânără.
Date de laborator. Nivelurile de IgG și IgA (la aproximativ 50% dintre pacienți) și IgM (până la cantități nedetectabile) sunt reduse semnificativ. Numărul de limfocite B din sânge este normal sau redus. Numărul de limfocite T la majoritatea pacienților este normal. La pacientii severi se poate dezvolta limfopenie (mai putin de 1500x103 celule in 1 litru de sange). Numărul de celule NK este redus. Producția de anticorpi specifici ca răspuns la imunizare este redusă sau absentă. Proliferarea limfocitelor și producția de IL-2 sub influența mitogenilor și antigenelor sunt afectate semnificativ.
Tabloul clinic. Sunt identificate boli infecțioase bacteriene recurente localizate în principal în tractul respirator și sinusurile paranazale. Până la momentul diagnosticului, infecțiile tractului respirator pot evolua spre bronșiectazie și leziuni difuze ale țesutului pulmonar. Posibilă infecție a sistemului digestiv, manifestată prin diaree, steatoree și malabsorbție (și, în consecință, pierderea greutății corporale). Infecții cauzate de Giardia lamblia, Pneumocystis carinii sau virusuri din familie Herpetoviridae. Pacienții cu CVID sunt predispuși la dezvoltarea artritei purulente cauzate de micoplasme și ureaplasme. Manifestările infecțiilor cu enterovirus pot fi encefalomielita, sindroame asemănătoare polimielitei și dermatomiozitei, leziuni ale pielii și mucoaselor. Autoimună Bolile sunt severe și pot determina prognosticul CVID. Uneori, primele manifestări clinice ale CVID sunt artrita, colita ulceroasă și boala Crohn, colangita sclerozantă, malabsorbția, LES, nefrita, miozita, boala pulmonară autoimună sub formă de pneumonită interstițială limfoidă, neutropenie,
purpură trombocitopenică, anemie hemolitică, anemie pernicioasă, alopecie totală, vasculită retiniană, fotosensibilitate. La pacienții cu CVID, frecvența (în 15% din cazuri) a granuloamelor asemănătoare sarcoidozei și a limfoproliferării non-maligne este semnificativ crescută. Tratament.
Chimioterapia antibacteriană.
Terapia de substituție: preparatele de imunoglobuline intravenoase se administrează la fiecare 3-4 săptămâni pe viață.
Pentru complicații autoimune, terapie imunosupresoare (glucocorticoizi, azatioprină, ciclosporină A) și posibilă prescripție de medicamente cu anticorpi monoclonali (infliximab etc.).
Sindroame hiper-IgM
Sindroamele hiper-IgM sunt boli destul de rare caracterizate printr-o scădere marcată sau absență completă a IgG, IgA și concentrații normale sau crescute de IgM seric. Acest lucru este cauzat de incapacitatea limfocitelor B de a efectua comutarea clasei de imunoglobuline și hipermutageneza domeniilor variabile. Până în prezent, au fost identificate 6 defecte genetice care duc la dezvoltarea sindromului hiper-IgM.
. Tip 1 (HIGM 1). Deficiența ligandului CD40 legat de X (70% din cazurile de sindroame hiper-IgM), ceea ce duce la incapacitatea celulelor T de a interacționa eficient cu limfocitele B.
. Tip 2 (HIGM 2). Autozomal recesiv, asociat cu un defect al AID - activarea indusă a citidin deaminazei (genă Aicda, 12р13)- o enzimă implicată în schimbarea clasei de imunoglobuline și hipermutageneză.
. Tip 3 (HIGM 3). Autozomal recesiv, asociat cu o mutație a genei moleculei CD40. În același timp, celulele B în sine nu sunt capabile să interacționeze eficient cu limfocitele T. Manifestările fenotipice sunt similare cu cele de tip 1.
. Tip 4 (HIGM 4). Autosomal recesiv; în unele cazuri apar mutaţii de novo. Asociat cu un defect al UNG - uracil-ADN glicozilaza, implicată și o enzimă
în schimbarea claselor de imunoglobuline, dar după acţiunea AID. În acest caz, hipermutageneza nu este afectată, iar sindromul apare cu mai puțină severitate.
. Tip 5 (HIGM 5). Defectul este doar în schimbarea clasei, hipermutageneza nu este afectată. Mutația cauzală nu a fost încă identificată, dar se pare că este un defect al enzimei care acționează după
AJUTOR.
. Tip 6 (HIGM-ED). Legat de X, asociat cu displazia ectodermală dishidrotică, este cauzat de deficiența NEMO (modulatorul NF-kB) care duce la afectarea semnalizării CD40.
sindromul hiper-IgM legat de X sunt detectate mai des decât altele. Se dezvoltă din cauza unui defect în gena care codifică CD40L (CD154, gena este situată pe Xq26-q27.2)- ligand pentru CD40. Expresia insuficientă a CD40L de către limfocitele T are ca rezultat eșecul comutării clasei de imunoglobuline în limfocitele B de la IgM la alte izotipuri, precum și afectarea formării celulelor B de memorie, a repertoriului celulelor T și a răspunsului celulelor Th1 direcționat împotriva microorganismelor intracelulare. . Băieții se îmbolnăvesc
Date de laborator. IgG, IgA, IgE nu pot fi determinate sau sunt detectate în cantități foarte mici. Nivelurile de IgM sunt normale (în 50% din cazuri) sau crescute, adesea semnificativ. Numărul de celule T și B este normal; răspunsul proliferativ al celulelor T indus de antigeni este redus. IgM sunt policlonale, uneori monoclonale. Sunt detectați autoanticorpi ai izotipului IgM (antieritrocitar, antiplachetar, antitiroidian, anticorpi la antigenele țesutului muscular neted). În țesutul limfoid nu există centri germinativi, dar există celule plasmatice.
Tabloul clinic. Primele manifestări apar în copilărie și copilărie timpurie. Se repetă infectii diverse localizări (în primul rând tractul respirator), inclusiv oportuniste (cauzate Pneumocystis carinii). Infecțiile virale sunt, de asemenea, tipice (citomegalovirus și adenovirusuri), Criptococcus neoformans, micoplasme și micobacterii. Infecția cu criptosporidiană poate provoca diaree acută și cronică (care se dezvoltă la 50% dintre pacienți) și colangită sclerozantă. Anemie, neutropenie, ulcerație a mucoasei bucale, gingivita, ulcerativ
leziuni ale esofagului, diferite părți ale intestinului, colită ulcerativă nespecifică. Identifică predispoziția la tulburări autoimune(artrita seronegativă, glomerulonefrită etc.) și neoplasme maligne (în principal țesut limfoid, ficat și tract biliar). Se pot dezvolta limfadenopatie, hepato- și splenomegalie. Tratament
Terapie regulată de înlocuire cu imunoglobulină intravenoasă.
Chimioterapia antibacteriană. Pentru prevenirea și tratamentul pneumoniei cu Pneumocystis se utilizează co-trimoxazol [sulfametoxazol + trimetoprim] și pentamidină.
Pentru a preveni afectarea ficatului și a căilor biliare, trebuie să beți numai apă fiartă sau filtrată și să efectuați examinări regulate (examinare cu ultrasunete, biopsie hepatică, dacă este indicat).
În tratamentul neutropeniei și al ulcerațiilor orale, se folosesc glucocorticoizi și preparate cu factor de stimulare a coloniilor de granulocite.
Odată cu dezvoltarea complicațiilor autoimune, este prescrisă terapia imunosupresoare (glucocorticoizi, azatioprină, ciclosporină A), precum și medicamente pe bază de anticorpi monoclonali.
Metoda optimă de tratament este transplantul de măduvă osoasă de la donatori potriviți cu HLA (rata de supraviețuire 68%, cel mai bine efectuat înainte de vârsta de 8 ani).
IMUNODEFICIENȚE COMBINATE CU DEFECT PREMINAR DE LIMFOCITUL T
Deficiență imunitară combinată severă
SCID Deficiență imunitară combinată severă)- un grup de sindroame caracterizate printr-o scădere a nivelului de limfocite T sau absența completă a acestora și o încălcare a imunității adaptive. . Disgeneza reticulară, caracterizată prin deteriorarea maturării precursorilor limfoizi și mieloizi în stadiile incipiente: neutropenie și T - B - NK - .
. SCID legat de X, rezultat dintr-o mutație genetică IL-2RG[(CD132, general la- lanț receptor pentru IL-2, IL-4, IL-7, IL-9, IL-15 și IL-21), Xq13.1-q21.1,אּ ], ceea ce duce la blocarea receptorilor și incapacitatea celulelor țintă de a răspunde la acțiunea interleukinelor corespunzătoare (mai mult de 50% din toate cazurile de SCID); T - B + NK - .
. Janus3 deficit de tirozin kinaza [genă JAK3 (19p13.1),ρ ]; cu defecte genice, transmiterea semnalului de activare de la general la- lanțuri de IL-2, IL-4, IL-7, IL-9, IL-15, IL-21 la nucleul celulei, ceea ce duce la diferențierea afectată a celulelor T și NK; T - B+NK - .
. Deficitul de proteina tirozin fosfatază (CD45, gena PTPRC, 1q31-q32); cu un defect genic, activitatea inhibitoare a kinazei Csk asupra proteinei tirozin kinazei Src este crescută cu fosforilarea afectată a domeniilor ITAM ale TCR și BCR; T - B+NK+.
. Deficiența completă a enzimelor RAG1 și RAG2, care activează recombinarea segmentelor V(D)J ale imunoglobulinelor și genelor TCR. RAG1Și RAG2 (11р13),ρ ]; T - B - NK + .
. Sindromul Omenn (deficit incomplet al enzimelor RAG1 și
RAG2) [gene RAG1și/sau RAG2 (11p13-p12),R]. Mulțumită
Datorită activității reziduale scăzute a acestor enzime, se dezvoltă un anumit număr de clone de limfocite T, specifice antigenelor țesuturilor epiteliale ale pielii și tractului digestiv, unde se înmulțesc și produc cantități mari de IL-4 și IL-5, provocând hipereozinofilie și formarea de IgE de către limfocitele B reziduale (în absența imunoglobulinelor din alte clase). Caracterizat prin eritrodermie și pahidermie cu alopecie la nivelul scalpului și sprâncenelor, diaree debilitantă, sindrom infecțios care pune viața în pericol; hepatosplenomegalie și hiperplazie ganglionară.
. SCID cu sensibilitate crescută la radiațiile ionizante. Defectul proteinei nucleare Artemis [genă DCLRE1C, (10p),R], parte a complexului de enzime necesare reparării ADN-ului (participă la îmbinarea rupurilor dublu-catene), când gena este mutată, recombinarea V(D)J este întreruptă; T - B - NK + .
. deficit de IL-2 [genă IL-2, 4q26-q27].
Mutații în gena lanțului a receptorului IL-2 (CD25) (10р15-р14);T - B + NK + .
Mutații în gena lanțului a receptorului IL-7 (CD127) (5р13);T - B + NK + .
deficiență TAP (Transportator pentru prezentarea antigenului), necesare pentru transportul peptidelor antigenice în reticulul endoplasmatic al genei lanțului a receptorului IL-7 (CD127) (5р13);T - B + NK + .
Mutații ale genelor lanțului CD3 (CD3γ, CDδ și CDε), conducând la scăderea numărului de limfocite T mature și diferențierea afectată; T - B + NK + .
Deficitul de protein tirozin kinaza ZAP-70 [genă ZAP-70 (2q12), R]. Când gena este mutată, este afectată fosforilarea domeniilor ITAM ale lanțului ζ TCR și receptorilor de celule NK care conțin ITAM și se dezvoltă o deficiență selectivă a celulelor T CD8 + (conținutul de limfocite T CD4 + este normal, dar funcțional). tulburările sunt exprimate sub forma unei lipse de producere a IL-de către aceste celule 2 și proliferare).
Deficitul de adenozin deaminază [genă ada (20q12-q13.11 , p)], conducând la acumularea de metaboliți în celule (deoxiadenozin trifosfat și S-adenosilhomocisteină) care inhibă proliferarea limfocitelor T și B (sunt descrise variante cu debut tardiv al bolii); T - B - NK - .
Deficitul de purin nucleozid fosforilază [genă pnp (14q11.2), p], ducând la acumularea de deoxiguanozin trifosfat în celule, care inhibă proliferarea limfocitelor T (sindroame asociate - uricemie și uricrie); T - B + NK - .
Date de laborator. Se detectează limfopenie variabilă, uneori profundă; limfocitele sunt incapabile să prolifereze ca răspuns la un antigen specific; Există adesea o scădere marcată a nivelului de imunoglobuline din serul sanguin. Nu există nicio umbră a timusului pe radiografia toracică.
Tabloul clinic. De obicei, diagnosticul clinic devine clar în primele 6 luni de viață, când dispar anticorpii IgG materni. În tabloul clinic, ele vin în prim-plan sindrom infecțios sever, hipoplazia țesutului limfoid și întârzierea dezvoltării. Sindromul infecțios se caracterizează prin candidoză orală, diaree cronică, pneumonie, febră,
sepsis de etiologie bacteriană, infecții virale. Agenții infecțioși aparțin diferitelor grupe taxonomice: bacterii, viruși, ciuperci, microorganisme oportuniste (Pneumocystis carinii). Pneumonia este adesea cauzată P. carinii, diaree - rotavirusuri, Campylobacter, Giardia lamblia. Hepatita virală se manifestă adesea. Dezvoltarea BCGitei regionale sau generalizate după vaccinare este tipică.
Tratament prevede prescrierea terapiei de întreținere, inclusiv nutriția parenterală, administrarea de imunoglobuline intravenoase, prescrierea de antibiotice, medicamente antifungice și antivirale. Una dintre principalele metode de tratament pentru a obține recuperarea este transplantul de măduvă osoasă, fără de care copiii cu SCID mor de obicei în primul an de viață. Au fost descrise cazuri izolate în care un copil, în condiții deosebit de igienizate, a trăit până la 2-3 ani. Este important să recunoaștem SCID la nou-născuți cât mai devreme posibil, deoarece imunizarea cu vaccinuri vii, de exemplu, este fatală pentru aceștia. Imediat după diagnostic, astfel de copii trebuie plasați în condiții gnotobiologice (cutie sterilă). În cazul bolilor infecțioase, se efectuează terapie intensivă antibacteriană, antivirală și antifungică și terapie de substituție cu imunoglobuline intravenoase. Pentru a preveni pneumonia pneumocystis, este prescris co-trimoxazol. În cazul dezvoltării BCG, este necesară terapia intensivă antituberculoasă pe termen lung. Pentru transfuzia componentelor sanguine, trebuie utilizate numai medicamente iradiate și filtrate. Există riscul de a dezvolta boală grefă contra gazdă post-transfuzie din cauza transferului transplacentar al limfocitelor materne.
Sindromul limfocitelor goale
Acesta este numele unei patologii când moleculele MHC-I sau MHC-II nu sunt exprimate în organism. În absența exprimării moleculelor MHC-I, conținutul de limfocite T CD8 + este redus și nu există activitate de celule NK; în absența MHC-II, nivelul limfocitelor T CD4 + este redus. Au fost caracterizate mai multe defecte genetice. Cu toate acestea, aceste defecte nu sunt localizate în genele MHC, ci în mai mulți factori diferiți responsabili pentru reglarea lor
expresie. Tabloul clinic sindromul limfocitelor goale și tratament similare celor pentru alte SCID.
sindromul DiGeorge
Sindromul DiGeorge sau sindromul defect al pungii faringiene a treia și a patra [deleții în 22q11, inclusiv gena TBX1 (22q11.2), relevă hipoplazia sau aplazia timusului, hipoplazia glandei paratiroide, defecte cardiace, deficit de limfocite T, număr variabil de limfocite B.
Date de laborator. O scădere semnificativă a numărului de celule T CD3+, CD4+ și CD8+ și o scădere bruscă a activității lor proliferative induse de mitogeni și antigeni. Numărul de celule B și NK este normal. Concentrațiile serice de imunoglobuline sunt în majoritatea cazurilor în limite normale; sunt posibile diferite variante de disgammaglobulinemie.
Tabloul clinic. Componenta imunodeficientei este reprezentata de hipoplazia sau aplazia timusului si recurenta, severa. boli infecțioase. Se detectează și hipoparatiroidismul (hipocalcemie și, ca urmare, tetanie, sesizabilă în zilele 1-2 după naștere); defecte ale sistemului circulator (inversarea dreaptă a arcului aortic, stenoza ventriculară dreaptă, defecte ale septurilor interventriculare și interatriale, tetralogia Fallot, atrezie sau hipoplazie a arterei pulmonare); cavitatea gurii; anomalii ale scheletului facial (distanță crescută între organele pereche, dimensiunea redusă a maxilarului, în special cea inferioară, urechi joase, filtru scurt). Anomalii severe în structura laringelui, faringelui, traheei, urechii interne, esofagului; tulburări de dezvoltare renală, sistemul nervos central și alte defecte de dezvoltare (polidactilie, absența unghiilor, atrezie anale, fistule anale). Caracterizat prin întârzierea vorbirii și dezvoltarea psihomotorie. Există o predispoziție la tulburări autoimune(citopenie, tiroidită autoimună) și neoplasme maligne.
Tratament.. Terapie antibacteriană și antivirală. . Terapie de substituție cu preparate de imunoglobuline intravenoase. . Tratament chirurgical pentru corectarea defectelor de dezvoltare. . Pentru complicații autoimune - terapie imunosupresoare. . În prezența endocrinopatiilor, corectarea tulburărilor corespunzătoare. . Transplantul de măduvă osoasă este ineficient
tivna. . Transplantul de țesut epitelial timic este justificat. Corectarea funcției glandei paratiroide.
Sindromul limfoproliferativ legat de X
X- sindromul limfoproliferativ legat este caracterizat printr-un răspuns imun afectat la virusul Epstein-Barr [cauzat de defecte genetice SH2D1A(SAP) în Xq25,אּ], ducând la proliferarea necontrolată a limfocitelor B transformate de virusul Epstein-Barr și infectarea de noi celule țintă de către virus.
Tabloul clinic. Au fost descrise cele mai frecvente 4 fenotipuri: mononucleoza infectioasa severa, afectiuni limfoproliferative maligne (limfoame, leucemii, in principal cu celule B), anemie sau pancitopenie (inclusiv datorita sindromului hemofagocitar indus de virus), disgamaglobulinemie. Infecția cu virusul Epstein-Barr este un mecanism de declanșare pentru formarea celor mai severe, rapid progresive și fatale boli: mononucleoza infecțioasă fulminantă (în 58% din cazuri duce la deces), sindromul hemofagocitar (fără tratament în 100% din cazuri duce la moarte). la moarte). În 10% din cazuri, fenotipul se manifestă înainte de infectarea cu virusul Epstein-Barr (în acest caz se dezvoltă de obicei disgammaglobulinemia și limfoamele). Cel mai adesea sunt identificate diferite tipuri de hipogammaglobulinemie. Imunodeficiența duce la dezvoltarea bacteriilor, fungice și virale boli infecțioase. Boala poate fi suspectată la băieții cu un istoric familial caracteristic și un test sero- sau PCR-pozitiv pentru virusul Epstein-Barr. Pentru diagnostic, se recomandă utilizarea unei combinații de analize genetice SH2D1Ași evaluarea nivelurilor de expresie SAP.
Tratament
În scopul prevenirii, se recomandă utilizarea medicamentelor antivirale - aciclovir, valaciclovir (administrarea lor precoce suprimă replicarea virusului Epstein-Barr în orofaringe) și imunoglobulină intravenoasă (cu un titru ridicat de anticorpi împotriva virusului Epstein-Barr). ).
Pentru hipogammaglobulinemie, imunoglobulina intravenoasă este utilizată lunar în combinație cu terapia antibacteriană.
Pentru tratamentul mononucleozei infecțioase fulminante, sunt prescrise doze mari de aciclovir și metilprednisolon, terapie cu doze mari cu imunoglobulină intravenoasă cu un titru ridicat de anticorpi împotriva virusului Epstein-Barr și IFN.
Când se dezvoltă sindromul hemofagocitar, dozele mari de dexametazonă sunt combinate cu Vepezid ♠ (etoposid).
La tratarea bolilor maligne se folosesc protocoale standard de terapie.
O metodă radicală de tratament este transplantul de măduvă osoasă de la donatori compatibili cu HLA.
Sindromul limfoproliferativ autoimun
Sindromul limfoproliferativ autoimun este un grup de boli caracterizate prin limfoproliferare benignă, hiperimunoglobulinemie, tulburări autoimune, creșterea limfocitelor CD3 + CD4 - CD8 - T (dublu negative) în sângele periferic și un defect de apoptoză [defect genetic Fas(CD95) - TNFRSF6 (10q24.1), gena caspaza-10, ligand Fas - FasL (1q23)].
Date de laborator. Conținutul de limfocite T CD3 + CD4 - CD8 - în sângele periferic sau în țesuturile limfoide este mai mare de 1%. Nivelurile de IgG, IgA și IgM pot fi normale, crescute sau chiar scăzute. Odată cu vârsta, hipergammaglobulinemia este înlocuită cu o concentrație scăzută de imunoglobuline serice, până la agammaglobulinemie. Sunt detectați autoanticorpi la eritrocite, trombocite, neutrofile, mușchi netezi și factor VIII; autoanticorpi antinucleari și antifosfolipidici, precum și factorul reumatoid etc. Limfocitoza este caracteristică.
Tabloul clinic. Toți pacienții au ficatul mărit, ganglionii limfatici (în primii 5 ani de viață) și splina. Limfoproliferarea nu este însoțită de febră și transpirații nocturne. Debut reacții autoimune poate să nu coincidă cu limfoproliferarea și să apară mai târziu. Odată cu vârsta, severitatea reacțiilor autoimune crește. Reacțiile autoimune împotriva celulelor sanguine (anemie hemolitică, trombocitopenie, neutropenie) se dezvoltă mai des, alte organe sunt mai rar afectate. Risc crescut de apariție a neoplasmelor maligne (limfoame T și B, limfom Burkitt, limfom atipic, limfogranulomatoză etc.).
Tratament.. Agenți chimioterapeutici (ciclofosfamidă, azatioprină, metotrexat, clorambucil). . Glucocorticoizi. . Splenectomie pentru hipersplenism sever și hemocitopenie. . În cazurile severe, este posibil transplantul de măduvă osoasă.
Sindromul de hiperimunoglobulinemie E
Sindromul hiper-IgE se caracterizează printr-o creștere semnificativă a nivelului de IgE seric, abcese repetate ale pielii și țesutului subcutanat de etiologie stafilococică, pneumonie cu formarea pneumocelului, anomalii în structura scheletului facial, AD. Natura genetică moleculară a sindromului hiper-IgE nu a fost încă stabilită. În unele cazuri, a fost dezvăluită moștenirea autosomal dominantă, în altele - moștenirea autosomal recesivă. Se presupune că defectele afectează moleculele de semnalizare ale receptorilor de citokine (în forma autosomal dominantă a acestui sindrom, mutații în Stat3)și poate fi asociat cu funcționarea afectată a subsetului de celule Th17. O altă genă responsabilă de formarea sindromului hiper-IgE este localizată pe cromozomul 4 (4q).
Date de laborator. Sunt detectate diverse tulburări imunologice: niveluri crescute de IgE în ser, chemotaxia neutrofilă afectată, formarea defectuoasă a anticorpilor; scăderea răspunsului HRT la anatoxinele candidină, difterice și tetanic; slăbirea activității proliferative a celulelor T ca răspuns la Candidași toxoid tetanic menținând în același timp răspunsul la mitogeni. Eozinofilie în sângele periferic și lichidul din abcesele pielii. Numărul de celule T și B este normal.
Tabloul clinic. Eczemă moderată la o vârstă fragedă. Trăsături caracteristice ale feței (punte lată a nasului, nas larg snub, asimetria scheletului facial, frunte proeminentă, ochi adânci, palat înalt). Ei detectează anomalii în dezvoltarea scheletului, scolioza, mobilitatea crescută a articulațiilor, tendința la fracturi osoase după leziuni minore și înlocuirea dentară afectată. Apar abcese ale pielii, țesutului subcutanat și ganglionilor limfatici. Pneumonia se dezvoltă la o vârstă mai înaintată (cei mai frecventi agenți patogeni sunt S. aureusȘi H. în-
gripa),în 77% din cazuri se formează un pneumocel, însoțit de o infecție cauzată de P. aeruginosaȘi A. fumigatus. Pneumonia poate apărea fără febră. Candidoza cronică a mucoaselor și unghiilor se dezvoltă în 83% din cazuri.
Tratament.. Terapie antibacteriană și antifungică pe termen lung (în scopul prevenirii - pe tot parcursul vieții). . Pentru a trata dermatita, se folosesc agenți topici, în cazuri severe - doze mici de ciclosporină A. Transplantul de măduvă osoasă este ineficient.
SIndroamele de defalcare cromozomiale
Pentru sindroame cu instabilitate cromozomiala: ataxiatelangiectazie[Defect al genei topoizomerazei ADN ATM (11q22), p] și sindromul Nijmegen[defect al genei nibrinei NBS1(8q21)] - caracterizată printr-o incidență crescută a tumorilor maligne, instabilitate cromozomială spontană și rupturi cromozomiale. Ambele proteine sunt implicate în repararea rupurilor ADN-ului dublu catenar și în reglarea ciclului celular. În mod normal, rupturile ADN-ului dublu catenar apar în timpul recombinării V(D)J a imunoglobulinei și a genelor TCR, schimbarea claselor de imunoglobuline, în timpul încrucișării și în timpul meiozei. Procese similare apar în timpul maturizării neuronilor creierului. Defecte în repararea ADN-ului în ataxia-telangiectazia și sindromul Nijmegen provoacă manifestări clinice, cum ar fi tulburări în sinteza imunoglobulinelor, a funcției organelor genitale și a sistemului nervos.
Ataxie-telangiectazie
Acest sindrom (frecvență 1:300 mii nou-născuți) cu un fenotip foarte eterogen a fost descris de medicul francez D. Louis-Bar. Simptomele ataxiei pot fi detectate la un copil încă de la vârsta de 2-4 luni. Ataxia este cauzată de degenerarea progresivă a celulelor Purkinje din cerebel. Telangiectaziile de pe pielea nasului, urechilor și conjunctivei apar ceva mai târziu, la vârsta de 3-6 ani. Pete de cafea cu lait apar adesea pe piele. Hipoplazia timusului, ganglionilor limfatici, splinei și amigdalelor este caracteristică. Imunodeficiența se manifestă printr-o scădere (adesea un dezechilibru) a producției de IgA, IgE, IgG2, IgG4. 80% dintre pacienți se dezvoltă
există simptome clinice infecțioase corespunzătoare. Numărul și activitatea funcțională a celulelor T (în principal celulelor T CD4 +) sunt reduse. Numărul total de limfocite T este normal la majoritatea pacienților. Incidența neoplasmelor (în principal limfoame și carcinoame) este neobișnuit de mare (de 200 de ori mai mare decât în populația generală), ducând adesea la deces până la vârsta de 10-12 ani. Tratament simptomatic.
sindromul Nijmegen
Sindromul Nijmegen (numit după orașul din Olanda unde a fost descrisă prima oară boala) se manifestă prin microcefalie, tulburări specifice ale scheletului facial (frunte înclinată, partea mijlocie proeminentă a feței, nas lung, hipoplazie a maxilarului inferior, forma ochiului mongoloid). , epicantus, urechi mari), dezvoltarea fizică întârziată, prezența petelor „café au lait” pe piele; clinodactilie și sindactilie, disgeneza ovariană etc. Majoritatea copiilor suferă de bacterii recurente și cronice. boli infecțioase tractul respirator, organele ORL și sistemul urinar. În 50% din cazuri se dezvoltă neoplasme maligne, predominant limfoame cu celule B. Sunt detectate diferite forme de disgamaglobulinemie și o scădere a celulelor T CD4 +.
Tratament.. Tratamentul simptomatic al tulburărilor neurologice. . Terapie de substituție cu imunoglobulină intravenoasă. . Terapia antibacteriană, antivirală și antifungică este utilizată conform indicațiilor. . La tratarea neoplasmelor maligne, se ia în considerare sensibilitatea crescută la radiații și chimioterapie.
Sindromul Wiskott-Aldrich
sindromul Wiskott-Aldrich [defect genetic WASP (Xp11.23p11.22),אּ; de asemenea ρ și Ʀ] Gen VIESPE(din sindromul Wiskott-Aldrich) exprimată în limfocite, țesutul splinei și timocite. Mutațiile acestei gene sunt asociate cu expresia anormală în neutrofile și limfocitele T (CD4 și CD8) ale moleculei CD43 (ligand pentru ICAM-1, îndeplinește o funcție anti-adeziv).
Date de laborator. Trombocitopenia (mai puțin de 10% din normal) este cauzată de distrugerea celulară crescută.
Trombocitele sunt mai mici decât cele ale oamenilor sănătoși. Nivelul de IgM din serul sanguin este redus cu un nivel normal de IgG și o creștere a conținutului de IgA și IgE. Titrurile izohemaglutininelor sunt reduse, formarea de anticorpi la antigenele polizaharide de pneumococ, streptococ, Escherichia coli, salmonella, precum și anticorpi antivirali este afectată. La o vârstă fragedă, de regulă, numărul de limfocite este normal; după 6 ani, se detectează limfopenie (mai puțin de 1x10 9 /l), o scădere a celulelor T CD3 + și CD4 + cu niveluri normale de celule B și NK. . Eozinofilia și dezvoltarea anemiei posthemoragice sunt posibile. Răspunsul proliferativ al celulelor T la mitogeni și antigene scade, iar HRT este slăbită. În splină nu sunt detectate structurile normale ale centrelor germinative și ale zonelor celulelor T.
Tabloul clinic. Boala se caracterizează printr-o triadă de simptome: trombocitopenie, eczemă și boli infecțioase recurente. Sindromul hemoragic se manifestă precoce, deja în perioada nou-născutului (erupție petehială, cefalohematoame, sângerare din rana ombilicală, sângerare intestinală). Eczema se manifestă de la o vârstă fragedă la 80% dintre pacienți. Odată cu vârsta, semnele imunodeficienței cresc: boli infecțioase bacteriene ale organelor ORL, ale sistemului respirator, ale organelor digestive, ale pielii; infecție herpetică răspândită sau generalizată (Herpes simplexȘi varicela zoster), citomegalovirus, precum și fungice (candidoza mucoaselor), mai rar boli infecțioase oportuniste. La 70% dintre pacienți sunt depistate boli autoimune (anemie hemolitică, neutropenie, artrită, vasculită cutanată, colită ulceroasă, vasculită cerebrală, glomerulonefrită, trombocitopenie autoimună). La pacienții cu vârsta peste 5 ani, frecvența este crescută neoplasme maligne(în principal tumori ale țesutului limfoid).
Tratament.. Transplant alogen de măduvă osoasă sau celule stem (rata de succes a operației ajunge la 90% când se folosește un transplant de la un donator histocompatibil și 50% când se folosește un transplant haploident). . Terapie de substituție cu imunoglobulină intravenoasă. . Administrarea preventivă de medicamente antibacteriene, antifungice și antivirale. . Pentru a reduce sindromul hemoragic, se efectuează splenectomia. . Pentru complicațiile autoimune este prescrisă terapia imunosupresoare.
DEFECTE ÎN FAGOCITOZE
Boala cronică granulomatoasă
Boala granulomatoasă cronică se caracterizează prin afectarea activității funcționale a fagocitelor (formarea de forme reactive de radicali de oxigen, distrugerea intracelulară și fragmentarea agenților patogeni fagocitați), boli infecțioase bacteriene și fungice persistente și dezvoltarea inflamației granulomatoase. Boala granulomatoasă cronică se dezvoltă la indivizi cu diferite defecte genetice [în 65% din cazuri - o variantă legată de X a bolii: gena gp91-phox (Xp21.1),אּ; în 35% din cazuri – autosomal recesiv: genă f47-phox (7q11.23),ρ; gena p67-phox (1q25),ρ; gena p22-phox (16q24), p], ducând la perturbări în sistemul NADP oxidază. Când neutrofilele de scurtă durată (câteva ore) mor, bacteriile neomorâte „se scurg” în locul inflamației. Macrofagele sunt celule cu viață lungă, iar precursorii lor (monocitele) migrează către leziune în cantități crescute (ceea ce duce la formare). granuloame), fagocitează microorganismele, dar nu sunt capabile să le omoare.
Date de laborator. Valorile normale ale imunoglobulinelor serice și ale subpopulațiilor de limfocite sunt caracteristice. Producția de radicali de peroxid de către neutrofile, evaluată în teste (chemiluminescență dependentă de luminol sau reducerea albastru de tetrazoliu), este brusc redusă sau absentă. Bolile infecțioase sunt caracterizate prin leucocitoză, neutrofilie, VSH crescut, anemie și hipergammaglobulinemie.
Tabloul clinic. Boala apare în majoritatea cazurilor în primul an de viață sindrom infectios(infecții cu agenți patogeni intra și extracelulari) și formarea de granuloame. Cele mai tipice sunt: afectarea plămânilor (pneumonie recurentă, afectarea ganglionilor limfatici hilari, abcese pulmonare, pleurezie purulentă), tractul digestiv, abcese cutanate și limfadenită. Cei mai frecventi agenți patogeni sunt microorganismele catalaze pozitive: S. aureus, Aspergillus spp., bacterii intestinale gram-negative (E. coli, Salmonella spp., Serratia marcescens), mai rar - Burkholderia cepaciaȘi Nocardia farcinica. Este caracteristică dezvoltarea abceselor hepatice și subdiafragmatice, a osteomielitei, a abceselor pararectale și a sepsisului.
Cea mai gravă complicație infecțioasă care pune viața în pericol este aspergiloza, care poate apărea sub formă de leziuni difuze ale plămânilor și ale altor organe (țesut adipos, creier, oase, articulații, endocard). Pacienții cu boală granulomatoasă cronică după vaccinarea BCG dezvoltă adesea o infecție asociată vaccinului care implică ganglionii limfatici regionali. Leziunile micobacteriene la pacienții cu boală granulomatoasă cronică pot avea atât localizare pulmonară, cât și extrapulmonară și au un curs prelungit. Pacienții cu boală granulomatoasă cronică se caracterizează printr-un întârziere în dezvoltarea fizică.
Tratament.. Terapie antimicrobiană: utilizarea profilactică continuă a co-trimoxazolului și a medicamentelor antifungice (itraconazol etc.); dacă apar complicații infecțioase se administrează parenteral terapia antibacteriană combinată (2-3 antibiotice bactericide care pătrund intracelular) în combinație cu terapia antifungică. Odată cu dezvoltarea aspergilozei, este indicată utilizarea pe termen lung a amfotericinei B sau a caspofunginei. Pentru infecțiile micobacteriene, se utilizează o combinație de terapie specifică pe termen lung cu medicamente antituberculoase și antibiotice cu spectru larg. . Tratamentul chirurgical este adesea însoțit de supurația plăgii postoperatorii și formarea de noi leziuni. Este posibilă drenajul prin puncție ghidat cu ultrasunete al abcesului. . Pentru a trata complicațiile infecțioase severe atunci când terapia antibacteriană este ineficientă, este posibil să se utilizeze masa de granulocite, doze mari de IFNu și G-CSF. . Transplantul de măduvă osoasă sau transplantul de celule sanguine din cordonul ombilical de la un frate compatibil poate avea succes la o vârstă fragedă, când riscul de deces din cauza complicațiilor infecțioase și a bolii grefă contra gazdă este minim.
Defecte de aderență a leucocitelor
Până în prezent, au fost descrise 3 defecte de adeziune a leucocitelor. Toate au
moștenire de tip autosomal recesiv, caracterizată prin boli infecțioase bacteriene și fungice repetate și cronice. Tipul I se caracterizează prin absența sau scăderea exprimării CD11/CD18 pe leucocite, chemotaxia neutrofilă afectată, leucocitoză (mai mult de 25x109), pierderea tardivă a cordonului ombilical și dezvoltarea omfalitei, vindecarea slabă a rănilor și lipsa formării de puroi. la locul pătrunderii agentului patogen în organism.
Tratament.. Terapie antibacteriană: episoade infecțioase și profilactice. . În cazurile severe, este prescris transplantul de măduvă osoasă de la un donator compatibil HLA.
DEFECTE ÎN SISTEMUL DE COMPLEMENT
Boli cu deficit de componente ale complementului
Manifestările defectelor genice ale componentelor individuale ale sistemului complement sunt prezentate în tabel. 11-2.
SA ereditar. Bolile cauzate de deficiența componentelor complementului sunt rareori detectate, deoarece manifestarea lor necesită o stare homozigotă pentru alelele autozomale. Există o excepție legată de C1inh (inhibitor al esterazei C1): mutația genei C1inh, conducând la deficit de inhibitor, în starea heterozigotă se manifestă printr-un fenotip cunoscut sub numele de AO ereditar (vezi capitolul 13, angioedem pentru mai multe detalii).
Boli imun complexe. Deficiența C1-C4 se manifestă prin dezvoltarea bolilor complexe imune - vasculită sistemică și afectarea rinichilor, care se numește colectiv sindromul lupus eritematos sistemic (LES).
Infecții piogene. Deficiența de C3 (și factorii H și I) este asociată cu o susceptibilitate crescută la infecții piogene. Deficiența componentelor implicate în calea alternativă de activare a complementului, precum și deficiența componentelor C5-C8, sunt asociate cu o susceptibilitate crescută la infecție cauzată de Neisseria spp. Deficitul de C9 este de obicei asimptomatic clinic.
Tabelul 11-2. Manifestări clinice ale defectelor componentelor individuale ale sistemului complement
Deficit de lectină care leagă manoza
Deficiența de lectină care leagă manoza (MBL) este cauzată de un defect al genei. MBL(diverse mutații punctuale și deleții ale genei MBL detectat la 17% dintre caucazieni). Cu defectele genelor, activarea proteazelor care descompun componentele complementului C2 și C4 și activarea sistemului complementului de-a lungul căii lectinei sunt perturbate. Din punct de vedere clinic această patologie se manifestă printr-un sindrom infecţios.
Date de laborator. Analiza subpopulațiilor de limfocite, leucocite și izotipuri de imunoglobuline nu arată abateri semnificative adecvate simptomelor clinice. Nu există MSL în serul de sânge.
Tratament. Această boală nu este o imunodeficiență clasică. Prin urmare, imunocorecția cu agenți imunotropi este contraindicată. MSL recombinant poate fi utilizat ca medicament farmacologic pentru terapia de substituție etiopatogenetică a pacienților cu acest defect ereditar. Acest medicament este în prezent în curs de studii clinice.
Hipogamaglobulinemie tranzitorie la copii
Hipogamaglobulinemia tranzitorie la copii este asociată cu o caracteristică fiziologică a formării treptate a sistemului de imunoglobuline. Maturarea formării anticorpilor IgM și IgA este „întârziată” în cea mai mare măsură. La copiii sănătoși, conținutul de IgG maternă scade treptat și după șase luni crește producția propriilor anticorpi IgG. La unii copii, cu toate acestea, creșterea nivelului de imunoglobuline este întârziată. Astfel de copii pot suferi de infecții bacteriene recurente. În aceste cazuri, nu trebuie să apelați la perfuzii de preparate de imunoglobuline donatoare (administrare intravenoasă de imunoglobuline).
Deficit selectiv de imunoglobulina A
Deficiența selectivă a imunoglobulinei A (SD IgA - Deficitul selectiv de IgA) se dezvoltă ca urmare a unui defect genetic tnfrsf13b
sau p). Deficiența de IgA în prezența imunoglobulinelor din alte clase este cea mai frecventă imunodeficiență detectată în populația generală cu o frecvență de 1:500-1500 de persoane (și mai des la pacienții cu alergii). Există deficit selectiv de IgA, adică constând într-o deficiență a uneia dintre subclase (30% din cazuri), și completă (70% din cazuri). Deficiența subclasei IgA2 duce la un tablou clinic mai sever decât deficiența subclasei IgA1. Sunt posibile și combinații ale deficitului de IgA cu alte tulburări: cu un defect în biosinteza IgG și cu anomalii ale limfocitelor T. Marea majoritate a oamenilor cu selectiv
Deficitul de IgA este practic sănătos. Pentru copiii sub 2 ani, deficitul de IgA este o afecțiune fiziologică.
Detectează o scădere a concentrației serice de IgA la<5 мг/дл у детей старше 4 лет; IgG и IgM в норме, количество и соотношение субпопуляций лимфоцитов и их функциональная активность могут быть в норме.
Tabloul clinic. Cu deficit de IgA se pot dezvolta 3 grupe de sindroame patologice: infecțioase, autoimune și alergice. Pacienții cu deficit de IgA sunt predispuși la infecții recurente ale tractului respirator superior și ale sistemului digestiv. Cele mai frecvente și severe sunt diverse boli autoimune (artrita reumatoidă, spondilita anchilozantă, sindromul Sjögren, vasculita cu afectarea vaselor cerebrale, tiroidita autoimună, LES, glomerulonefrita, anemie hemolitică, diabet zaharat tip I, vitiligo etc.). Incidența bolii celiace este de 10 ori mai mare decât la copiii cu IgA normale. Manifestările alergice cel mai frecvent identificate sunt: intoleranța la proteinele din laptele de vacă, dermatita atopică (DA), astmul bronșic.
Tratament. Cazurile asimptomatice nu necesită tratament special; în prezența manifestărilor clinice ale bolilor infecțioase, autoimune și alergice, tratamentul se efectuează în conformitate cu standardele.
Terapia de substituție cu imunoglobuline de la donator nu este indicată nici pentru deficiența selectivă sau completă de IgA, deoarece există o probabilitate mare ca primitorul să dezvolte anticorpi anti-izotipici la IgA și să dezvolte complicații de transfuzie cauzate de aceștia.
Agamaglobulinemie cu deficit de celule B
Agammaglobulinemie legată de X (boala lui Bruton) reprezintă 90% din toate cazurile de agammaglobulinemie. Sunt afectați băieții și fiii (אּ, ρ) purtători ai genei defecte btk (Xq21.3-q22), care codifică proteina tirozin kinaza specifică limfocitelor B Btk (tirozin kinaza lui Bruton- tirozin kinaza lui Bruton). Ca urmare a defectului, căile de semnalizare intracelulară sunt întrerupte, recombinarea lanțurilor grele de imunoglobuline și
transferul celulelor pre-B în limfocitele B. La 10% dintre pacienți, agammaglobulinemia cu deficit de celule B este moștenită autosomal recesiv. În prezent, au fost descrise 6 defecte genetice, inclusiv moleculele receptorului celulelor pre-B, proteina adaptoare a celulelor B citoplasmatice (BLNK) și gena Repetă bogată în leucină care conține 8 (LRRC8).
Date de laborator. Nu există limfocite B periferice. Măduva osoasă conține celule pre-B cu lanțul μ în citoplasmă. Numărările celulelor T și testele funcționale ale celulelor T pot fi normale. IgM și IgA nu pot fi detectate în sânge; IgG poate fi prezentă, dar în cantități mici (0,4-1,0 g/l). Nu există anticorpi la antigenele grupelor de sânge și antigenele de vaccin (tetanos, toxine difterice etc.). Se poate dezvolta neutropenie. Examinarea histologică a țesutului limfoid: nu există centre germinale (germinale) și celule plasmatice în foliculii limfoizi.
Tabloul clinic. Dacă antecedentele familiale sunt necunoscute, diagnosticul devine evident la vârsta medie de 3,5 ani. Boala se caracterizează prin hipoplazie a țesutului limfoid, infecții purulente severe, boli infecțioase ale tractului respirator superior (sinuzită, otită) și inferioară (bronșită, pneumonie); posibil gastroenterită, piodermie, artrită septică (bacteriană sau chlamydia), septicemie, meningită, encefalită, osteomielita. Cei mai frecventi agenți patogeni care provoacă boli ale tractului respirator sunt Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus, diaree bacterii intestinale sau Giardia Giardia lamblia. De asemenea, pacienții cu agammaglobulinemie sunt susceptibili la boli infecțioase cauzate de micoplasme și ureaplasme, care provoacă dezvoltarea pneumoniei cronice, artritei purulente, cistita și abcese ale țesutului subcutanat. Virușii tipici sunt virusurile neurotropice ECHO-19 și coxsackie, care provoacă atât encefalită acută și cronică severă, cât și encefalomielita. Manifestările infecțiilor cu enterovirus pot include sindrom asemănător dermatomiozitei, ataxie, dureri de cap și tulburări de comportament. La copiii bolnavi, atunci când sunt imunizați cu vaccinul poliomielitei viu, de regulă, este detectată eliberarea prelungită a virusului poliomielitei prin membranele mucoase, cu virulența restabilită și în creștere (adică în colectivele de copii).
există un pericol real ca copiii sănătoși să se infecteze cu poliomielita ca urmare a contactului cu un copil imunodeficient vaccinat). Tulburările autoimune în agammaglobulinemie pot fi reprezentate de poliartrita reumatoidă, sindromul asemănător sclerodermiei, scleredemul, colita ulceroasă, diabetul zaharat tip I (datorită predominării răspunsului imun Th1).
Examinare fizică. Atenție la întârzierea dezvoltării fizice, la forma degetelor (degete sub formă de tobe), modificări ale formei toracelui, caracteristice bolilor tractului respirator inferior, hipoplazie a ganglionilor limfatici și amigdalelor.
Tratament.
Terapia de substituție: preparatele de imunoglobuline intravenoase se administrează la fiecare 3-4 săptămâni pe viață. Dozele de imunoglobuline sunt selectate astfel încât să creeze o concentrație în serul pacientului care depășește limita inferioară a normei de vârstă.
Discutarea posibilității terapiei genice - genă Btk clonat, dar supraexpresia sa este asociată cu transformarea malignă a țesutului hematopoietic.
În cazul neutropeniei persistente se folosesc factori de creștere. Dacă apar semne de patologie autoimună, se pot prescrie medicamente cu anticorpi monoclonali (infliximab etc.).
Deficiență imunitară variabilă comună
Deficiența imună variabilă comună (CVID) este un grup de sindroame caracterizate printr-un defect în sinteza anticorpilor și a imunității celulare. Un criteriu de diagnostic fiabil pentru CVID este o scădere semnificativă a conținutului de imunoglobuline a două sau trei izotipuri principale la persoanele de ambele sexe în combinație cu unul dintre următoarele simptome:
Debutul bolii este peste 2 ani;
Lipsa izohemaglutininelor și/sau răspuns scăzut la vaccinare;
Excluzând alte cauze ale agammaglobulinemiei.
La unii pacienți, cauza dezvoltării CVID este mutațiile în genele care codifică moleculele implicate în procesele de maturare și supraviețuire a celulelor B: BAFF-R (Receptorul factorului de activare al celulelor B), Blimp-1 (proteina de maturare indusă de limfocitele B-1)și ICOS (Costimulator inductibil). Există o întrerupere a capacității limfocitelor B de a se diferenția în celule plasmatice, se dezvoltă defecte în formarea anticorpilor, este posibilă disfuncția limfocitelor T și se observă o susceptibilitate crescută la boli infecțioase. Sindromul poate apărea în copilărie timpurie, adolescență sau vârsta adultă tânără.
Date de laborator. Nivelurile de IgG și IgA (la aproximativ 50% dintre pacienți) și IgM (până la cantități nedetectabile) sunt reduse semnificativ. Numărul de limfocite B din sânge este normal sau redus. Numărul de limfocite T la majoritatea pacienților este normal. La pacientii severi se poate dezvolta limfopenie (mai putin de 1500x103 celule in 1 litru de sange). Numărul de celule NK este redus. Producția de anticorpi specifici ca răspuns la imunizare este redusă sau absentă. Proliferarea limfocitelor și producția de IL-2 sub influența mitogenilor și antigenelor sunt afectate semnificativ.
Tabloul clinic. Sunt identificate boli infecțioase bacteriene recurente localizate în principal în tractul respirator și sinusurile paranazale. Până la momentul diagnosticului, infecțiile tractului respirator pot evolua spre bronșiectazie și leziuni difuze ale țesutului pulmonar. Posibilă infecție a sistemului digestiv, manifestată prin diaree, steatoree și malabsorbție (și, în consecință, pierderea greutății corporale). Infecții cauzate de Giardia lamblia, Pneumocystis carinii sau virusuri din familie Herpetoviridae. Pacienții cu CVID sunt predispuși la dezvoltarea artritei purulente cauzate de micoplasme și ureaplasme. Manifestările infecțiilor cu enterovirus pot fi encefalomielita, sindroame asemănătoare polimielitei și dermatomiozitei, leziuni ale pielii și mucoaselor. Autoimună Bolile sunt severe și pot determina prognosticul CVID. Uneori, primele manifestări clinice ale CVID sunt artrita, colita ulceroasă și boala Crohn, colangita sclerozantă, malabsorbția, LES, nefrita, miozita, boala pulmonară autoimună sub formă de pneumonită interstițială limfoidă, neutropenie,
purpură trombocitopenică, anemie hemolitică, anemie pernicioasă, alopecie totală, vasculită retiniană, fotosensibilitate. La pacienţii cu CVID, frecvenţa de neoplasme maligne(în 15% din cazuri), granuloame asemănătoare sarcoidozei și limfoproliferare non-malignă. Tratament.
Chimioterapia antibacteriană.
Terapia de substituție: preparatele de imunoglobuline intravenoase se administrează la fiecare 3-4 săptămâni pe viață.
Pentru complicații autoimune, terapie imunosupresoare (glucocorticoizi, azatioprină, ciclosporină A) și posibilă prescripție de medicamente cu anticorpi monoclonali (infliximab etc.).
Sindroame hiper-IgM
Sindroamele hiper-IgM sunt boli destul de rare caracterizate printr-o scădere marcată sau absență completă a IgG, IgA și concentrații normale sau crescute de IgM seric. Acest lucru este cauzat de incapacitatea limfocitelor B de a efectua comutarea clasei de imunoglobuline și hipermutageneza domeniilor variabile. Până în prezent, au fost identificate 6 defecte genetice care duc la dezvoltarea sindromului hiper-IgM.
. Tip 1 (HIGM 1). Deficiența ligandului CD40 legat de X (70% din cazurile de sindroame hiper-IgM), ceea ce duce la incapacitatea celulelor T de a interacționa eficient cu limfocitele B.
. Tip 2 (HIGM 2). Autozomal recesiv, asociat cu un defect al AID - activarea indusă a citidin deaminazei (genă Aicda, 12р13)- o enzimă implicată în schimbarea clasei de imunoglobuline și hipermutageneză.
. Tip 3 (HIGM 3). Autozomal recesiv, asociat cu o mutație a genei moleculei CD40. În același timp, celulele B în sine nu sunt capabile să interacționeze eficient cu limfocitele T. Manifestările fenotipice sunt similare cu cele de tip 1.
. Tip 4 (HIGM 4). Autosomal recesiv; în unele cazuri apar mutaţii de novo. Asociat cu un defect al UNG - uracil-ADN glicozilaza, implicată și o enzimă
în schimbarea claselor de imunoglobuline, dar după acţiunea AID. În acest caz, hipermutageneza nu este afectată, iar sindromul apare cu mai puțină severitate.
. Tip 5 (HIGM 5). Defectul este doar în schimbarea clasei, hipermutageneza nu este afectată. Mutația cauzală nu a fost încă identificată, dar se pare că este un defect al enzimei care acționează după
. Tip 6 (HIGM-ED). Legat de X, asociat cu displazia ectodermală dishidrotică, este cauzat de deficiența NEMO (modulatorul NF-kB) care duce la afectarea semnalizării CD40.
sindromul hiper-IgM legat de X sunt detectate mai des decât altele. Se dezvoltă din cauza unui defect în gena care codifică CD40L (CD154, gena este situată pe Xq26-q27.2)- ligand pentru CD40. Expresia insuficientă a CD40L de către limfocitele T are ca rezultat eșecul comutării clasei de imunoglobuline în limfocitele B de la IgM la alte izotipuri, precum și afectarea formării celulelor B de memorie, a repertoriului celulelor T și a răspunsului celulelor Th1 direcționat împotriva microorganismelor intracelulare. . Băieții se îmbolnăvesc
Date de laborator. IgG, IgA, IgE nu pot fi determinate sau sunt detectate în cantități foarte mici. Nivelurile de IgM sunt normale (în 50% din cazuri) sau crescute, adesea semnificativ. Numărul de celule T și B este normal; răspunsul proliferativ al celulelor T indus de antigeni este redus. IgM sunt policlonale, uneori monoclonale. Sunt detectați autoanticorpi ai izotipului IgM (antieritrocitar, antiplachetar, antitiroidian, anticorpi la antigenele țesutului muscular neted). În țesutul limfoid nu există centri germinativi, dar există celule plasmatice.
Tabloul clinic. Primele manifestări apar în copilărie și copilărie timpurie. Se repetă infectii diverse localizări (în primul rând tractul respirator), inclusiv oportuniste (cauzate Pneumocystis carinii). Infecțiile virale sunt, de asemenea, tipice (citomegalovirus și adenovirusuri), Criptococcus neoformans, micoplasme și micobacterii. Infecția cu criptosporidiană poate provoca diaree acută și cronică (care se dezvoltă la 50% dintre pacienți) și colangită sclerozantă. Anemie, neutropenie, ulcerație a mucoasei bucale, gingivita, ulcerativ
leziuni ale esofagului, diferite părți ale intestinului, colită ulcerativă nespecifică. Identifică predispoziția la tulburări autoimune(artrita seronegativă, glomerulonefrită etc.) și neoplasme maligne (în principal țesut limfoid, ficat și tract biliar). Se pot dezvolta limfadenopatie, hepato- și splenomegalie. Tratament
Terapie regulată de înlocuire cu imunoglobulină intravenoasă.
Chimioterapia antibacteriană. Pentru prevenirea și tratamentul pneumoniei cu Pneumocystis se utilizează co-trimoxazol [sulfametoxazol + trimetoprim] și pentamidină.
Pentru a preveni afectarea ficatului și a căilor biliare, trebuie să beți numai apă fiartă sau filtrată și să efectuați examinări regulate (examinare cu ultrasunete, biopsie hepatică, dacă este indicat).
În tratamentul neutropeniei și al ulcerațiilor orale, se folosesc glucocorticoizi și preparate cu factor de stimulare a coloniilor de granulocite.
Odată cu dezvoltarea complicațiilor autoimune, este prescrisă terapia imunosupresoare (glucocorticoizi, azatioprină, ciclosporină A), precum și medicamente pe bază de anticorpi monoclonali.
Metoda optimă de tratament este transplantul de măduvă osoasă de la donatori potriviți cu HLA (rata de supraviețuire 68%, cel mai bine efectuat înainte de vârsta de 8 ani).
Aceasta este cea mai frecventă imunodeficiență primară apare cu o frecventa de 1/300 - 1/700. Pentru prima dată a existat...
descris de J. Heremans în 1960.
În cele mai multe cazuri, nu există un model clar de moștenire a bolii; în cazuri mai rare de pierdere familială a IgA, moștenirea autosomal recesivă este mai frecventă decât moștenirea dominantă. Deficitul de IgA este uneori asociat cu anomalii ale cromozomului 18.
IgA diferă de alte imunoglobuline prin conținutul de carbohidrați și acizi sialici și prin capacitatea de a forma dimeri, trimeri și chiar tetrameri. Există 2 variante de molecule de IgA:
— IgA serică este întotdeauna un monomer;
- IgA secretorie - formeaza di-, tri- si tetrameri inainte de a deveni parte integranta a secretiilor (saliva, lacrimi, lapte, secretii ale cailor digestive, respiratorii si urinare).
Un lanț proteic special, lanțul J, participă la formarea IgA secretoare. Prin urmare, acesta este un întreg sistem de proteine. De aici formele patologiei IgA:
- deficitul general de IgA este asociat cu anomalii în sinteza monomerului IgA. Ca urmare, conținutul atât de IgA ser, cât și de IgA secretorie este redus. Sunt încălcate atât protecția locală, cât și cea generală;
- defect în formarea moleculelor secretoare - SIgA. Motivul poate fi absența lanțului J, care duce la afectarea imunității locale;
- o încălcare predominantă a sintezei IgA serice, când sunt secretate numai forme secretoare de IgA și practic nu sunt secretați monomeri ai IgA serice.
Simptomele clinice depind de gradul de deficit de IgAși se pot manifesta sub formă de patologie a organelor și sistemelor individuale. Cele mai frecvente sunt leziunile infecțioase ale membranelor mucoase.
Din tractul gastrointestinal - gastrită cronică, colită ulceroasă și hemoragică, ileită, stomatită. Din partea sistemului respirator - rinită, sinuzită, bronșită, care devine rapid cronică. Bronhopneumonia cronică duce adesea la bronșiectazie și abcese pulmonare. S-a constatat, de asemenea, o legătură între un nivel scăzut al IgA secretoare și o incidență crescută a alergiilor atopice și dezvoltarea bolilor autoimune (lupus eritematos sistemic, poliartrită reumatoidă, tiroidită autoimună).
La persoanele cu un deficit selectiv de IgA monomerică predominant seric, nu apar cel mai adesea manifestări clinice ale imunodeficienței.
Cercetare de laborator: arată că numărul total de limfocite T și B din sângele periferic al unor astfel de pacienți este în limite normale. Nivelurile serice și/sau secretorii de IgA sunt reduse brusc, în timp ce nivelurile de IgM și IgG sunt în limite normale. La unii pacienți (cu manifestări de alergii atopice), nivelurile IgE pot fi crescute.
Tratament: simptomatic. Trebuie să fiți atenți atunci când utilizați terapia de înlocuire cu imunoglobuline, deoarece la administrarea repetată există riscul de șoc anafilactic din cauza posibilității de creștere a producției de antiimunoglobuline din clasele IgG și IgM.
Prognoza: Cursul bolii este în principal benign.
Criteriu de diagnostic- scăderea la pacienții cu vârsta peste 4 ani a nivelurilor serice de IgA mai mici de 0,07 g/l cu niveluri normale de IgG și IgM și excluderea altor cauze de hipogamaglobulinemie. Semnificativ din punct de vedere diagnostic:
O scădere izolată a nivelului de IgA în ser (mai puțin de 0,05 g/l) cu niveluri normale ale altor izotipuri de imunoglobuline la copiii cu vârsta peste 1 an, absența IgAl și IgA2. Nivelurile de IgM și IgG sunt normale. Cu toate acestea, unii pacienți au deficit de IgG2;
- dacă nivelul IgA este în intervalul de la 0,05 g/l până la 0,2 g/l, atunci este diagnosticat deficit parțial de IgA; numărul normal de limfocite T și subclasele lor;
- de obicei un număr normal de limfocite B (CD19\CD20);
- număr normal de celule NK (CD16 CD56).
La pacienţii cu deficit de IgA, mai ales în absența IgA secretoare, este necesar să se examineze nivelul subclaselor de IgA. La unii pacienți, deficitul selectiv de IgA poate progresa spre dezvoltarea ulterioară a CVID. Este necesară monitorizarea regulată pe termen lung a nivelurilor de imunoglobuline (inclusiv la pacienții asimptomatici).
Determinarea autoanticorpilor (antinucleari, antitiroidieni etc.).
Cu mancare intoleranţă sau malabsorbția, testarea alergiilor și determinarea anticorpilor la lapte și a anticorpilor IgG anti-gluten.
Tratament. Pacienții cu deficit selectiv de IgA asimptomatic nu necesită tratament continuu. Pacienților cu manifestări de boli infecțioase li se prescriu antibiotice în scop profilactic. Tratamentul antibacterian intensiv se efectuează la toți pacienții în timpul apariției unei boli infecțioase. Pacienții nu sunt contraindicați pentru imunizarea de rutină. Terapia de substituție cu imunoglobuline este contraindicată atunci când pacientul are autoanticorpi împotriva IgA. Trebuie luat în considerare faptul că deficitul selectiv de IgA este un defect imun primar necorectabil. Măsurile terapeutice se limitează la tratamentul simptomatic al bolilor infecțioase, alergice și autoimune. Medicamentele imunotrope sunt prescrise în principal din cauza manifestării morbidității infecțioase crescute.
Prognoza. La pacienții cu deficit selectiv de IgA, prognosticul depinde de prezența unui defect concomitent în anticorpi specifici, alergii sau boli autoimune. Adesea, cursul asimptomatic al bolii poate fi perturbat din cauza acțiunii factorilor externi dăunători, de exemplu, într-o situație stresantă, imunosupresie, chimioterapie etc.

