Selektyvus IgA trūkumas yra dažniausias pirminis imunodeficito sutrikimas (PIDS). Pacientų, sergančių selektyviu IgA trūkumu, dažnis svyruoja nuo 1: 400 iki 1: 1000 kaukaziečių populiacijoje ir daug mažesnis, nuo 1: 4000 iki 1: 20 000, mongoloidu. Jungtinėse Amerikos Valstijose ligos paplitimas svyruoja nuo 1 iš 223-1000 tirtoje grupėje iki 1 iš 400-3000 sveikų kraujo donorų. Tokių tyrimų Rusijoje nebuvo atlikta.
Šiai būklei būdingas selektyvus IgA koncentracijos sumažėjimas serume, mažesnis nei 0,05 g / l (vyresniems kaip ketverių metų vaikams), normalus lygis kiti imunoglobulinai serume, normali reakcija antikūnų serume ir normalų ląstelių sukeltą imuninį atsaką. Daugumoje tyrimų dažnis tarp vyrų ir moterų buvo maždaug vienodas.
Žmonės, negalintys gaminti IgA, gali kompensaciniais mechanizmais toleruoti savo ligą asimptomiškai arba kenčia nuo dažnų kvėpavimo, virškinimo ar urogenitalinė sistema, gastroenterologinė patologija (pavyzdžiui, celiakija), polinkis į atopinius sutrikimus, tokius kaip šienligė, bronchų astma, atopinis dermatitas, tarpininkaujama IgE maisto alergija, taip pat neurologinės ir autoimuninės ligos (dažniausiai tai reumatoidinis artritas, sisteminė raudonoji vilkligė, idiopatinė trombocitopeninė purpura, Sjogreno sindromas). Su selektyviu IgA trūkumu alerginės ligos, tokių kaip atopinis dermatitas ir bronchinė astma, pasitaikė 40% atvejų (Consilium Medicum, 2006). Anafilaksinės reakcijos perpilant kraujo komponentus ir skiriant į veną imunoglobulinus, taip pat būdingos daugumai šių pacientų, kurios yra susijusios su IgA buvimu šiuose produktuose.
Klinikiniai selektyvaus IgA trūkumo simptomai gali pasireikšti ankstyvoje vaikystėje, o perduodamų infekcijų dažnis ir sunkumas su amžiumi gali mažėti dėl kompensacinio IgG1 ir G3, IgM poklasio antikūnų padidėjimo. Kitas nebuvimo paaiškinimas klinikiniai simptomai nepaisant sumažėjusio imunoglobulino kiekio serume, gali būti normalus sekrecijos IgA lygis. Arba, priešingai, kai kuriems pacientams, kuriems iš pradžių diagnozuotas selektyvus IgA trūkumas, gali pasireikšti bendro kintamo imuninio trūkumo klinikinė išraiška.
Selektyvaus IgA trūkumo terapija šiuo metu yra gretutinių ligų nustatymas, vedimas prevencinės priemonėsopiumas siekiant sumažinti infekcijos riziką, taip pat greitai ir efektyviai gydyti infekcijas.
Specifinio gydymo nėra. Pacientų, sergančių IgA trūkumu, prognozė paprastai yra gera, jei nėra ryškių klinikinės apraiškos... Laikui bėgant, vaikų IgA trūkumas gali būti papildytas.
Būdamos genetiškai nustatytos, imunodeficito būsenos atsiranda dėl genetinio aparato defektų. Pacientai, turintys bendrą kintamą imuninį trūkumą, ir tie, kuriems yra selektyvus IgA trūkumas, dažnai būna toje pačioje šeimoje ir turi bendrą HLA haplotipą; daugelis turi retus alelius ir genų ištrynimus MCH klasėje - 3 chromosomos 6 klasėje. Neseniai įrodyta, kad kai kuriuos šeiminius įprasto kintamo imuninio deficito ir selektyvaus IgA trūkumo atvejus sukelia TNFRSF13B geno mutacijos, koduojančios baltymą, žinomą kaip TACI (transmembraninis aktyvatorius ir kalcio moduliatorius ir ciklofilino-ligando sąveika). Tikėtina, kad tais atvejais, kai TACI mutacijos nebuvo aptiktos, ligos priežastis gali būti spontaniškos ar paveldimos kitų genų mutacijos, kurios dar nėra užfiksuotos.
Šiuo metu pakankamai išsamiai aprašomos galimos selektyvaus IgA trūkumo klinikinės apraiškos, eigos variantai ir galimos gretutinės ligos. Lemiamas diagnozuojant ligą yra selektyvus IgA koncentracijos serume sumažėjimas vaikams nuo 4 metų iki 0,05 g / l, esant normalioms kitų serumo imunoglobulinų koncentracijoms pakartotinėse imunogramose. Gydymas yra gretutinių ligų nustatymas, prevencinių priemonių taikymas siekiant sumažinti infekcijos riziką, taip pat būtina greitai ir veiksmingas gydymas užkrečiamos ligos.
Nėra informacijos apie šios pirminės imunodeficito būklės pasireiškimo dažnumą Rusijos gyventojams, todėl neįmanoma palyginti ligos paplitimo mūsų šalyje su kitomis šalimis, kuriose panašūs tyrimai jau buvo atlikti.
Pagrindinė problema yra vienodų rekomendacijų dėl pacientų, turinčių selektyvų IgA trūkumą, valdymo taktikos trūkumas.
Siekiant įvertinti selektyvaus IgA trūkumo pasireiškimo dažnumą tarp grupės vaikų ambulatorinis stebėjimas "Dažnai sergantys vaikai" ir apibūdinkite jo klinikinių apraiškų diapazoną Rusijos Federacija Rusijos Federacijos Sveikatos apsaugos ministerijos FGBU „Dmitrijaus Rogachevo vardu pavadinto FNKTS DGOI“ ir 9 vardu pavadinto GBUZ DGKB 9 pagrindu. G. N. Speransky DZM šis darbas buvo atliktas.
Medžiagos ir tyrimo metodai
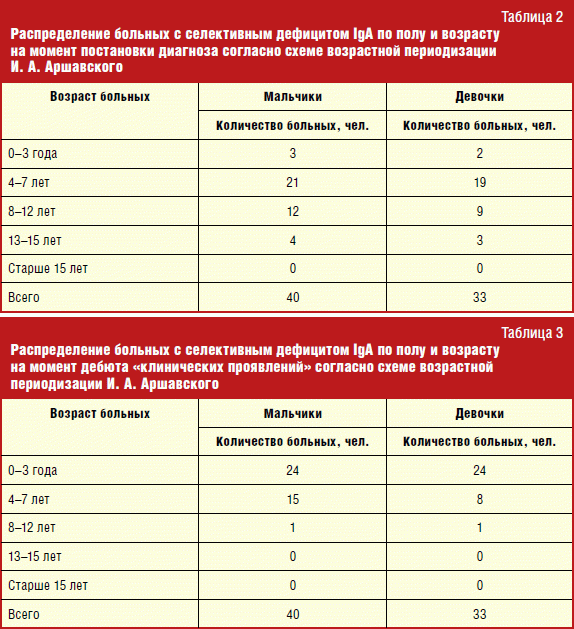
Tyrimo objektas buvo vaikai, turintys selektyvų IgA trūkumą, kurie buvo pastebėti GBUZ DGKB Nr. 9 pavadintame G.N.Speranskio DZM. Be to, buvo atlikta retrospektyvi medicinos dokumentų analizė už laikotarpį nuo 2003 iki 2010 m. 9154 pacientai iš ambulatorinio stebėjimo grupės „dažnai sergantys vaikai“ (1-3 lentelės).
Tyrimo metu buvo naudojami šie metodai:
- klinikiniai ir anamneziniai;
- generolas ir biocheminės analizės kraujas;
- imunologinis kraujo sudėties tyrimas nefelometrijos ir srauto citometrijos metodais;
- skarifikavimo bandymai;
- specifinio IgE nustatymas imunobloto būdu;
- išorinio kvėpavimo funkcijos tyrimas;
- rinocitologinis tyrimas.
Selektyvaus IgA trūkumo diagnozė buvo nustatyta remiantis selektyviu IgA koncentracijos sumažėjimu serume žemiau 0,05 g / l normalus veikimas kiti imunoglobulinai serume pakartotinėse imunogramose ir kitų išskyrimas galimos priežastys jų trūkumas vyresniems nei 4 metų vaikams.
Vartojant anamnezę ypatingas dėmesys buvo pateiktas klinikinių pasireiškimų dažnis ir spektras, gretutinė patologijair šeimos istorija buvo išsamiai išnagrinėta. Klinikinis vaikų tyrimas buvo atliekamas pagal visuotinai priimtus metodus. A, G, M, E klasių imunoglobulinų kiekis serume buvo nustatytas nefelometriškai naudojant BN 100 nefelometrą (Dade Bering, Vokietija) naudojant Dade Behring rinkinį. Limfocitų fenotipai buvo nustatyti srauto citometrija naudojant „FacsScan“ įrenginį (Becton Dickenson, JAV), naudojant fluorescenciškai pažymėtus monokloninius antikūnus „Simultest“ (Becton Dickenson, JAV). Pacientai, turintys bet kokių atopijos apraiškų, taip pat visi pacientai, sergantys pakeltas lygis IgE, kuris buvo nustatytas atlikus imuninės būklės rodiklių vertinimą nefelometrijos būdu, buvo atliktas tolesniam alerginiam tyrimui, atliekant skarifikavimo tyrimus vyresniems nei 4 metų vaikams arba specifinio IgE nustatymo serume metodą pacientams iki 4 metų. Vaikams, kuriems nustatyta bronchinė astma arba kurie sirgo bronchų obstrukciniu sindromu, buvo atliktas išorinio kvėpavimo funkcijos tyrimas naudojant Spirovit SP-1 aparatą (Schiller AG, Šveicarija). Taip pat buvo atlikti visi reikalingi papildomi susijusių specialistų tyrimai ir konsultacijos, atsižvelgiant į esamus skundus.
Rezultatai ir jų aptarimas
Retrospektyvi pacientų, kuriems siunčiamos diagnozės „pasikartojantis ARVI“, „CWD“, „CBS“, taip pat „EBD“, medicininių dokumentų analizė leido nustatyti, kad šios grupės vaikų selektyvaus IgA trūkumo dažnis yra du ar net tris kartus didesnis nei gyventojų.
Absoliutų skaičių, taip pat vaikų, sergančių šiuo pirminiu imunodeficitu, procentą pagal metus galima pamatyti lentelėje. 4.
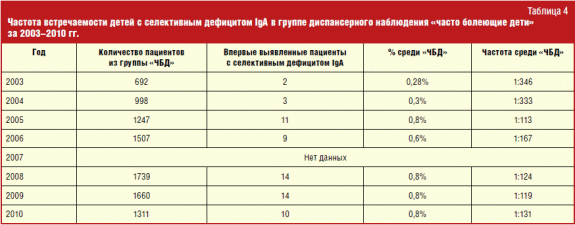
Deja, 2007 m. Duomenų nėra. 2003 ir 2004 m. Buvo konsultuojamasi su 692 ir 998 vaikais. Tarp jų iš viso buvo nustatyti 5 pacientai, turintys selektyvų IgA trūkumą, o tai yra šiek tiek dažniau nei vidutinis gyventojų skaičius - atitinkamai 1: 346 ir 1: 333, palyginti su 1: 400-600. Nuo 2005 m. Naujai diagnozuotų pacientų, sergančių šia PIDS, dažnis labai išaugo: 1: 113 2005 m., 1: 167 2006 m., 1: 124 2008 m., 1: 119 2009 m. Ir galiausiai , 1: 131 2010 m. Tyrimo metu įvykių dažnis pasikeitė nuo 1: 346 2003 m. Iki 1: 131 2010 m., Kai jis pasirodė didžiausias, palyginti su ankstesniais metais. Pacientų, sergančių selektyviu IgA trūkumu, padidėjimas trečiaisiais metais nuo darbo pradžios turėtų būti siejamas su padidėjusiu gydytojų budrumu šios patologijos atžvilgiu, taip pat su pagerėjimu. laboratorinė diagnostika... Būtina toliau plėsti gydytojų žinias apie šią ligą, nes vaikų, kurių tėvai atneša pas imunologą su skundais, srautas dažnos ligos, kasmet didėja.
Atliekant šį darbą perspektyviai taip pat buvo tiriami 235 vaikai ir 32 suaugusieji.
Pagrindinę grupę sudarė 73 vaikai, kuriems diagnozuotas selektyvus IgA trūkumas.
Antroje pacientų grupėje buvo 153 vaikai, sergantys idiopatine trombocitopenine purpura (ITP). Pacientų, sergančių ITP, imuninės būklės įvertinimas buvo atliktas siekiant nustatyti tarp jų selektyvų IgA trūkumą, nes ši koreliacija aprašyta pasaulinėje literatūroje ir tie patys duomenys buvo gauti Šis tyrimas... Tarp jų neradome nė vieno vaiko, kuriame nebūtų IgA. Nepaisant to, kad, tiriant ITP sergančių pacientų imuninę būklę, nepavyko nustatyti selektyvaus IgA trūkumo tarp jų, buvo nustatyti kiti nedideli humoraliniai defektai: IgG poklasių trūkumas, kūdikių hipogammaglobulinemija, dalinis IgA sumažėjimas.
Trečiajai grupei priklausė 32 suaugusieji nuo 20 iki 54 metų, taip pat 8 vaikai nuo 4 iki 10 metų, kurie yra artimiausi selektyvaus IgA trūkumo pacientų giminaičiai, kuriems buvo atliktas imuninės būklės įvertinimas, siekiant surasti ir apibūdinti šeimos atvejus.
Apklausos ir gautų duomenų analizės metu gauti šie rezultatai.
Vyrų ir moterų santykis tarp pacientų, sergančių selektyviu IgA trūkumu, buvo maždaug toks pat. Buvo ištirta 40 berniukų ir 33 mergaitės. Tai atitinka pasaulinės literatūros duomenis.
Didžiausias selektyvaus IgA trūkumo aptikimas buvo 4–7 metų amžiaus. Pakartotinės infekcinės ligos, kaip taisyklė, pasitaikė ankstyvas amžius arba prasidėjus vizitui ikimokyklinėje įstaigoje. Paprastai prieš patekdami pas imunologą vaikai sukaupė tam tikrą infekcinę istoriją, nes yra tam tikrų požymių, leidžiančių įtarti PIDS. Be to, net jei tyrimas buvo atliktas ankstesniame amžiuje ir IgA nebuvimas buvo atskleistas iki 4 metų, tai neleido mums nustatyti nedviprasmiškos PIDS diagnozės, mes negalėjome visiškai atmesti imunoglobulino sintezės sistemos nebrandumo. Todėl iki 4 metų diagnozė buvo nustatyta remiantis klausimais ir rekomenduota imtis tolesnių veiksmų. Taigi atitinkamai 4–7 metų intervalas.
Pagrindiniai skundai dėl vaikų, turinčių selektyvų IgA trūkumą, buvo dažni kvėpavimo takai virusinės infekcijos su nesudėtingu kursu. Pasikartojančių kvėpavimo takų ligų debiutas, kaip taisyklė, buvo 3 metų amžiaus. Tai taip pat atitinka pasaulinės literatūros duomenis. Kadangi dinaminė daugumos mūsų tyrimo pacientų kontrolė buvo vykdoma ilgai, keletą metų, kartais prieš paciento perėjimą prie suaugusiųjų tinklo, galima teigti, kad perduodamų infekcijų dažnis ir sunkumas mažėjo su amžiumi. Tikėtina, kad tai lėmė kompensacinis IgG1 ir IgG3, IgM poklasių antikūnų padidėjimas, tačiau šiam klausimui reikia papildomų tyrimų. Antras dažniausias skundas gydymo metu buvo dažnos ūminės kvėpavimo takų virusinės infekcijos su komplikacijomis. Mūsų pacientų komplikuotų, netipinių ARVI su amžiumi dažnis, kaip rodo dinaminis stebėjimas, taip pat sumažėjo.
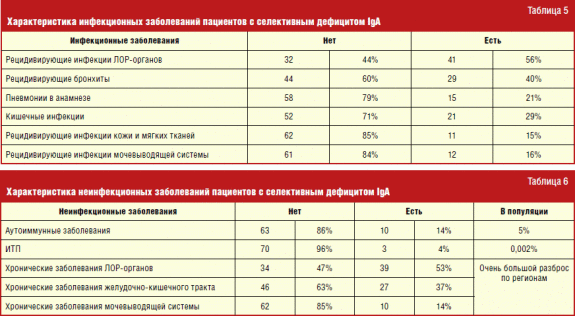
Tarp infekcinių ligų spektro pacientams, kuriems yra selektyvus IgA trūkumas, ENT organų infekcinės ligos ir apatinės dalies infekcijos kvėpavimo takai... Taip yra dėl to, kad sumažėjęs sekrecinis IgA, kuris yra vietinio imuniteto dalis, sukelia lengvą infekciją ir mikroorganizmų dauginimąsi ant gleivinės, kurios yra labiausiai pažeidžiamos kontaktuojant su užkrečiamos ligosperduodama oro lašeliniu būdu.
Neužkrečiamųjų ligų spektre buvo nustatyta akivaizdi koreliacija su autoimuninėmis ligomis, kurios yra svarbiausios selektyvaus IgA trūkumo apraiškos, ypač su idiopatine trombocitopenine purpura (1,5–2 100 tūkst.).
Apie autoimuninės ligos pacientams, sergantiems selektyviu IgA trūkumu, dažniausiai pasireiškė jaunatvinis reumatoidinis artritas (4 kartus), lėtinė idiopatinė trombocitopeninė purpura (3 kartus) ir autoimuninis hepatitas (3 kartus). Be to, remiantis pasauline literatūra, pacientams, kuriems yra selektyvus IgA trūkumas, tarp artimiausių giminaičių dažniau pasitaiko autoimuninių ligų. Tačiau, remiantis mūsų tyrimais, jų skaičius neviršijo bendros gyventojų vertės.
Atopinių ligų dažnis tarp pacientų, sergančių selektyviu IgA trūkumu, buvo žymiai didesnis nei populiacijoje (4 lentelė). Tik alerginio rinito dažnis yra panašus į bendrą populiaciją. Tokie pastebėjimai atsispindi daugelyje ankstesnių tyrimų. Negalima sakyti, kad daugumai pacientų, kuriems yra IgA trūkumas, alerginės ligos yra sunkesnės nei žmonėms, neturintiems šio imunologinio defekto. Tačiau dėl didelio atopijos paplitimo kyla klausimas dėl imunologinio tyrimo atlikimo siekiant nustatyti selektyvaus IgA trūkumo formas, kurios dar kliniškai nepasireiškė. Nors tai gali neturėti lemiamo vaidmens požiūriu į gydymą esant dabartinei atopinei būklei, tai padės laiku diagnozuoti ir sumažinti galimą riziką žmonėms, kuriems bus nustatytas selektyvus IgA trūkumas.
Analizuojant pakartotines imunogramas dinaminio stebėjimo metu vaikams, turintiems selektyvų IgA trūkumą, dėl nuolatinių laboratorinių parametrų pokyčių didelės grupės pacientų. A grupė parodė IgA nebuvimą be jokių kitų pokyčių. Grupėje be IgA jis buvo derinamas su nuolatiniu IgG lygio padidėjimu. Buvo atlikta lyginamoji šių pacientų grupių analizė.

Klinikinių pasireiškimų atsiradimo amžius šiose grupėse reikšmingai nesiskyrė.
Nustatyta, kad pacientams, kuriems yra selektyvus IgA trūkumas, IgG lygio padidėjimas koreliuoja su pasikartojančiomis odos ir minkštųjų audinių infekcijomis. Šį klausimą reikia toliau nagrinėti.
Lyginant šias pacientų grupes, reikšmingų alerginės patologijos spektro skirtumų nenustatyta.
Darbo metu imuninė būklė buvo įvertinta 20 pacientų, sergančių selektyviu IgA trūkumu, šeimose. Buvo nustatyti 4 šeimos atvejai. Be to, buvo surinkta išsami šeimos istorija. Tarp suaugusių giminaičių, turinčių užkrautą infekcinę istoriją, galėjusių atlikti tyrimą, buvo tam tikrų humoralinio imuniteto pažeidimų. Atitinkamai, nustačius nedidelius humoralinius defektus (ypač selektyvų IgA trūkumą), artimiausių giminaičių tyrimas, ypač esant užkrautai infekcinei anamnezei, yra privalomas.
Atsižvelgiant į tai, kad ambulatorinės stebėjimo grupės vaikų „dažnai sergantys vaikai“ selektyvus IgA trūkumas yra daug dažnesnis nei bendroje vaikų populiacijoje, praktikuojantys pediatrai turi būti atsargūs dėl šios ligos. Ne visada lengva tai įtarti, nes klinikinės apraiškos yra labai įvairios: nuo besimptomių formų iki pasikartojančių bakterinių infekcijų, kai reikia dažnai antibakterinė terapija... Rekomenduojama išplėsti pediatrų ir siaurų ambulatorinės ir stacionarinės priežiūros specialistų žinias apie nedidelius humoralinio imuniteto defektus.
Kadangi tarp pacientų, sergančių selektyviu IgA trūkumu, alerginės patologijos (bronchinės astmos, atopinio dermatito, alergijos maistui) dažnis yra žymiai didesnis, autoimuninių ir hematologinių ligų dažnis, taip pat lėtinės ligos (ENT organai, urogenitalinė sistema, virškinimo trakto), nei populiacijoje, ją identifikuoti būtina, kad būtų galima pateikti išsamią ir savalaikę informaciją medicininė priežiūra pacientų.
Vaikus, kurie anksčiau sirgo infekcinėmis ligomis, pacientus, sergančius hematologinėmis ir autoimuninėmis ligomis, rekomenduojama nukreipti pas imunologą / atlikti imunologinį tyrimą, taip pat atlikti viso IgA lygio atrankinį tyrimą pacientams, sergantiems alerginėmis ligomis.
Tyrimas parodė, kad dauguma vaikų, turinčių selektyvų IgA trūkumą, parodė koreliaciją tarp autoimuninė patologija ir nuolatinis IgG padidėjimas pakartotinėse imunogramose. Sergant kitomis ligomis, tokia koreliacija nebuvo nustatyta. Tokie rodiklių pokyčiai yra vaiko autoimuninės patologijos vystymosi rizikos veiksnys ir reikalauja ypatingo dėmesio.
Nepaisant to, kad koreliacija tarp selektyvaus IgA trūkumo šeimos anamnezės ir pacientų klinikinių pasireiškimų sunkumo nebuvo nustatyta, šiems pacientams artimiausių giminaičių tyrimas, ypač esant užkrautai infekcinei anamnezei, yra privalomas.
Literatūra
- Hammarstrom L., Lonnqvist B., Ringden O., Smith C. I., Wiebe T. IgA trūkumo perdavimas kaulų čiulpams persodintam pacientui, kuriam yra aplastinė anemija // Lancet. 1985 m. 1 (8432): 778-781.
- Latiff A. H., Kerr M. A.Klinikinė imunoglobulino A trūkumo reikšmė // Klinikinės biochemijos metraščiai. 2007 m. 44 (Pt 2): 131-139.
- Al-Attas R. A., Rahi A. H.Pirminis antikūnų trūkumas arabams: pirmoji ataskaita iš rytų Saudo Arabijos // Journal of Clinical Immunology. 1998; 18 (5): 368-371.
- Carneiro-Sampaio M. M., Carbonare S. B., Rozentraub R. B., de Araujo M. N., Riberiro M. A., Porto M. H.Selektyvaus IgA trūkumo dažnis tarp Brazilijos kraujo donorų ir sveikų nėščių moterų // Allergology Immunopathology (Madr). 1989; 17 (4): 213-216.
- Ezeoke A. C. Selektyvus IgA trūkumas (SIgAD) Rytų Nigerijoje // African Journal of Medicine and Medical Sciences. 1988 m. 17 (1): 17–21.
- Feng L. Epidemiologinis selektyvaus IgA trūkumo tyrimas tarp 6 tautybių Kinijoje // Zhonghua Yi Xue Za Zhi. 1992 m. 72 (2): 88-90, 128.
- Pereira L. F., Sapina A. M., Arroyo J., Vinuelas J., Bardaji R. M., Prieto L. Selektyvaus IgA trūkumo paplitimas Ispanijoje: daugiau, nei mes manėme // Kraujas. 1997; 90 (2): 893.
- Wiebe V., Helal A., Lefranc M. P., Lefranc G. Molekulinė T17 imunoglobulino CH daugelio genų ištrynimo analizė (del A1-GP-G2-G4-E) // Žmogaus genetika. 1994; 93 (5): 5.
L.A. Fedorova *,
E. S. Puškova *
I. A. Korsunsky **, 1, Medicinos mokslų kandidatas
A.P. Prodeus *,medicinos mokslų daktaras, profesorius
Imunodeficitas - pagrindinių komponentų kiekybinių rodiklių ir (arba) funkcinio aktyvumo sumažėjimas imuninė sistema, dėl kurio pažeidžiama kūno apsauga nuo patogeninių mikroorganizmų ir pasireiškia padidėjęs infekcinis sergamumas.
Kaip žinote, pagrindinė imuninės sistemos funkcija yra antigeninio pobūdžio pašalinių medžiagų, kurios prasiskverbia į kūną, atpažinimas ir pašalinimas aplinka (mikroorganizmai) arba atsiranda endogeniškai (naviko ląstelės). Ši funkcija įgyvendinama naudojant veiksnius įgimtas imunitetas (fagocitozė, antimikrobiniai peptidai, komplemento sistemos baltymai, NK ląstelių sistema ir kt.) ir įgytas arba prisitaikantis imunitetas, atliekamas naudojant ląstelių ir humoralinius imuninius atsakus. Organizmo imuninės gynybos komponentų aktyvumo ir jų sąveikos reguliavimas vyksta citokinų ir tarpląstelinių kontaktų pagalba.
Kiekviename iš išvardytų imuninės sistemos komponentų, taip pat jų reguliavimo mechanizmuose, gali atsirasti sutrikimų, dėl kurių išsivysto imunodeficitas, kurio pagrindinis klinikinis pasireiškimas yra padidėjęs jautrumas infekcinių ligų sukėlėjams. Yra 2 imunodeficito tipai: pirminis ir antrinis.
Pirminiai imunodeficitas(PID) - paveldimos ligossukeltų imuninį atsaką kontroliuojančių genų defektų. PID - įvairaus pobūdžio ir imuninių defektų, klinikinių apraiškų ir molekulinių sutrikimų ligos. Klinikiniam PID vaizdui būdingi pasikartojantys ir lėtiniai, sunkūs infekciniai procesai, daugiausia bronchopulmoninė sistema.
ir ENT organai, oda ir gleivinės; Gali išsivystyti pūlingas limfadenitas, abscesai, osteomielitas, meningitas ir sepsis. Tam tikromis formomis pasireiškia alergijos, autoimuninės ligos ir kai kurių vystymasis piktybiniai navikai... Turėtumėte atkreipti dėmesį į atsilikimą amžiaus rodikliai fizinis vystymasis... Šiuo metu aprašyta apie 80 PID ir nustatyti genai, atsakingi už daugumos šių ligų vystymąsi. Atitinkami laboratoriniai tyrimai leidžia diferencijuoti patologiją limfocitų lygyje ir patologiją ne limfocitinių antigenų sunaikinimo ir pašalinimo mechanizmų lygiu.
PID paplitimaspriklauso nuo ligos formos ir vidutiniškai svyruoja nuo 1:10 000 iki 1: 100 000 naujagimių. Selektyvus trūkumas Pavyzdžiui, IgA yra daug labiau paplitęs nuo 1: 500 iki 1: 1500 žmonių. Paplitimas skirtingų formų PID skiriasi skirtingos salys... Dažniausi antikūnų gamybos defektai - 50-60% atvejų, kartu PID - 10-30%, fagocitozės defektai - 10-20%, komplemento defektai - 1-6%. Dauguma PID pasireiškia ankstyvoje vaikystėje, nors vėliau gali pasireikšti kai kurios PID formos, ypač įprastas kintamasis imunologinis trūkumas (CVID).
Pagal plėtros mechanizmus yra 4 pagrindinės PID grupės:
1-oji grupė - daugiausia humoralinė, arba B-ląstelė
PID;
2 grupė - kombinuoti PID (su visais T ląstelių imunodeficitais yra B ląstelių disfunkcija);
3-oji grupė - PID, kurią sukelia fagocitozės defektai;
4-oji grupė - PID, kurį sukelia komplemento sistemos defektai.
Pirminio imunodeficito diagnozavimo principai
Anksti diagnozavus ir laiku pradėjus gydymą, nustatoma ligos prognozė. Nustatant diagnozę rajono pediatrų lygiu, kyla tam tikrų sunkumų, kurie dažnai kyla dėl to, kad nėra galimybės laiku konsultuoti pacientą su imunologu ir atlikti specialų laboratorinį imunologinį tyrimą (11-1 lentelė). Nors žinios apie PID klinikinio vaizdo ypatybes ir pokyčiai
atliekant klinikinius laboratorinius tyrimus, galima įtarti PID ir nukreipti pacientą pas specialistus. Europos imunodeficito draugija sukūrė ankstyvosios PID diagnostikos protokolus ir taip pat sukūrė elektroninė bazė duomenys iš Europos PID registro. PID diagnostikos algoritmas parodytas fig. 11-1.
11-1 lentelė.Imunologinio tyrimo etapai su įtariamu imunodeficitu
Paveikslėlis: 11-1.Pirminio imunodeficito diagnostikos algoritmas
Pagrindiniai pirminio imunodeficito klinikinio vaizdo ypatumai
Vedantis klinikinis vaizdas PID yra vadinamasis infekcinis sindromas - padidėjęs jautrumas infekcinių ligų sukėlėjams apskritai, neįprastai sunkus pasikartojantis (pasikartojantis) klinikinė eiga, netipinių ligos sukėlėjų (dažnai oportunistinių) buvimas ligos etiologijoje. Patogeno tipą lemia imuninio defekto pobūdis. Esant antikūnų gamybos defektams, galima nustatyti atsparumą antibakteriniai vaistai flora - stafilokokai, streptokokai, Haemophilus influenzae. Esant T ląstelių imuniniam trūkumui, be bakterijų, nustatomi virusai (pavyzdžiui, herpeso virusų šeima), grybai (Candida spp., Aspergillusir kiti), o su fagocitiniais defektais - stafilokokai, gramneigiamos bakterijos, grybai ir kt.
Laboratoriniai tyrimai
Jei klinikiniai duomenys rodo PID, reikia atlikti šiuos tyrimus:
Išsamaus kraujo kiekio nustatymas (ypač svarbūs limfocitų kiekybiniai ir procentiniai rodikliai);
IgG, IgA ir IgM kiekio serume nustatymas;
T ir B limfocitų pogrupių skaičiavimas;
◊ fagocitų funkcinės būklės analizė (paprasčiausia ir informatyviausia analizė yra tetrazolio mėlynojo atstatymo testas);
Complement pagrindinių komplemento komponentų turinio analizė (pradėkite nuo C3 ir C4);
◊ ŽIV infekcijos tyrimas (jei yra galimų rizikos veiksnių);
◊ molekuliniai genetiniai tyrimai, kai nurodoma.
Pirminio imunodeficito gydymo principai
Pagrindinis PID terapijos tikslas yra gydyti ir užkirsti kelią ligos komplikacijoms. Šis požiūris yra susijęs su tuo, kad PID imuninės sistemos defektai yra genetiniame lygyje. Šiuo metu atliekami intensyvūs genų tyrimai
imunodeficito terapija, dėl kurios gali atsirasti radikalesni jų gydymo metodai.
Priklausomai nuo PID formos, gydymą sudaro pakaitinė terapija, infekcinių, autoimuninių ligos pasireiškimų gydymas ir prevencija, piktybinių navikų gydymas ir specialūs metodai, įskaitant kraujodaros kamieninių ląstelių transplantaciją (atsižvelgiant į PID tipą).
IMUNOGLOBULININIAI DEFEKTAI
Vaikų laikina hipogammaglobulinemija
Vaikų laikina hipogammaglobulinemija yra susijusi su fiziologiniu imunoglobulino sistemos fazinio formavimosi ypatumu. Labiausiai „vėluoja“ IgM ir IgA antikūnų susidarymas. Sveikiems vaikams motinos IgG kiekis palaipsniui mažėja, o po šešių mėnesių padidėja jų pačių IgG antikūnų gamyba. Tačiau kai kurių vaikų imunoglobulino koncentracijos padidėjimas vėluoja. Tokie vaikai gali sirgti pasikartojančiomis bakterinėmis infekcinėmis ligomis. Tokiais atvejais donoro imunoglobulino infuzijos (į veną leidžiamo imunoglobulino) vartoti negalima.
Selektyvus imunoglobulino A trūkumas
Selektyvus imunoglobulino A (SD IgA - Selektyvus IgA trūkumas)išsivysto dėl genų defekto tnfrsf13b
arba p). IgA trūkumas, esant kitų klasių imunoglobulinams, yra dažniausias imunodeficitas, nustatytas visoje populiacijoje, kurio dažnis yra 1: 500-1500 žmonių (dar dažniau alergiškiems pacientams). Atskirkite selektyvų IgA trūkumą, t.y. susideda iš vieno poklasio trūkumo (30% atvejų) ir visiško (70% atvejų). IgA2 poklasio trūkumas lemia ryškesnį klinikinį vaizdą nei IgA1 poklasio trūkumas. Taip pat galimi IgA trūkumo deriniai su kitais sutrikimais: su IgG biosintezės defektu ir su T-limfocitų anomalijomis. Didžioji dauguma žmonių, turinčių selektyvų
igA trūkumas yra praktiškai sveikas. Vaikams iki 2 metų IgA trūkumas yra fiziologinė būklė.
IgA koncentracijos serume sumažėjimas iki<5 мг/дл у детей старше 4 лет; IgG и IgM в норме, количество и соотношение субпопуляций лимфоцитов и их функциональная активность могут быть в норме.
Klinikinis vaizdas.Esant IgA trūkumui, gali išsivystyti 3 patologinių sindromų grupės: infekciniai, autoimuniniai ir alerginiai. Pacientai, turintys IgA trūkumą, yra linkę pasikartoti viršutinių kvėpavimo takų ir virškinimo sistemos infekcijose. Dažniausios ir sunkiausios yra įvairios autoimuninės ligos (reumatoidinis artritas, ankilozuojantis spondilitas, Sjogreno sindromas, vaskulitas su smegenų kraujagyslių pažeidimu, autoimuninis tiroiditas, SLE, glomerulonefritas, hemolizinė anemija, I tipo cukrinis diabetas, vitiligo ir kt.) Celiakijos dažnis yra 10 kartų didesnis nei vaikams, kurių IgA yra normali. Dažniausiai nustatomos alerginės apraiškos: karvės pieno baltymų netoleravimas, atopinis dermatitas (AD), bronchinė astma.
Gydymas.Asimptominiams atvejams nereikia specialaus gydymo; esant infekcinių, autoimuninių ir alerginių ligų klinikinėms apraiškoms, gydymas atliekamas pagal standartus.
Pakaitinė terapija donoriniais imunoglobulinais nenurodyta nei esant selektyviam, nei visiškam IgA trūkumui, nes yra didelė tikimybė, kad recipiente susidarys anti-izotipiniai antikūnai prieš IgA ir atsiras jų sukeltų perpylimo komplikacijų.
Agammaglobulinemija su B ląstelių trūkumu
Su X susijusi agammaglobulinemija (Brutono liga)sudaro 90% visų agammaglobulinemijos atvejų. Serga defektinio geno nešiotojai, berniukai, sūnūs (אּ, ρ) bTK (Xq21.3-q22),koduojantis B limfocitams būdingą baltymų tirozino kinazę Btk (Brutono tirozino kinazė- Brutono tirozino kinazė). Dėl defekto pažeidžiami tarpląsteliniai signalizacijos keliai, sunkiųjų imunoglobulinų grandinių rekombinacija,
pre-B ląstelių perteikimas B-limfocitams. 10% pacientų agammaglobulinemija su B ląstelių trūkumu paveldima autosominiu recesyviniu būdu. Šiuo metu aprašyti 6 genetiniai defektai, įskaitant prieš B ląstelių receptorių, citoplazminį B ląstelių adapterio baltymą (BLNK) ir geną Kartotinai turintis leucino, kuriame yra 8 (LRRC8).
Laboratorinių tyrimų duomenys.Periferinių B limfocitų nėra. Kaulų čiulpuose yra prieš B ląstelių, kurių citoplazmoje yra μ grandinė. T-limfocitų skaičius ir T-limfocitų funkcijos tyrimai gali būti normalūs. IgM ir IgA kraujyje nustatyti negalima; IgG gali būti, tačiau mažais kiekiais (0,4–1,0 g / l). Antikūnų prieš kraujo grupių antigenus ir vakcinos antigenus (stabligės, difterijos toksinų ir kt.) Nėra. Gali išsivystyti neutropenija. Histologinis limfoidinio audinio tyrimas: limfoidiniuose folikuluose nėra gemalo (embriono) centrų ir plazmos ląstelių.
Klinikinis vaizdas.Jei nežinoma šeimos istorija, diagnozė paaiškėja vidutiniškai 3,5 metų. Ligai būdinga limfoidinio audinio hipoplazija, sunkios pūlingos infekcijos, viršutinių (sinusito, vidurinės ausies uždegimo) ir apatinių (bronchito, plaučių uždegimo) kvėpavimo takų infekcinės ligos; galimas gastroenteritas, pioderma, septinis artritas (bakterinis ar chlamidinis), septicemija, meningitas, encefalitas, osteomielitas. Kvėpavimo ligų sukėlėjai dažniausiai yra Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus,viduriavimas žarnyno bakterijos arba lamblia Giardia lamblia.Taip pat pacientai, sergantys agammaglobulinemija, yra linkę į mikoplazmų ir ureaplazmų sukeltas infekcines ligas, kurios yra lėtinės pneumonijos, pūlingo artrito, cistito ir poodinio audinio abscesų atsiradimo priežastis. Tipiški virusai yra neurotropiniai virusai ECHO-19 ir coxsackie, kurie sukelia ir sunkų ūminį, ir lėtinį encefalitą, ir encefalomielitą. Enterovirusinių infekcijų apraiška gali būti į dermatomiozitą panašus sindromas, ataksija, galvos skausmai ir elgesio sutrikimai. Sergantiems vaikams, skiepijant gyva poliomielito vakcina, paprastai nustatomas ilgalaikis poliomielito viruso išsiskyrimas per gleivinę ir atkurtas bei didėjantis virulentiškumas (t. Y.
yra realus sveikų vaikų, užsikrėtusių poliomielitu, užkrėtimo pavojus dėl kontakto su vakcinuotu imunodeficito vaiku). Autoimuniniai sutrikimai sergant agammaglobulinemija gali pasireikšti reumatoidiniu artritu, į sklerodermą panašiu sindromu, skleredema, opiniu kolitu, I tipo cukriniu diabetu (dėl vyraujančio Th1 imuninio atsako).
Medicininė apžiūra.Atkreipkite dėmesį į fizinio vystymosi atsilikimą, į pirštų formą (pirštai būgnelių pavidalu), krūtinės formos pokyčius, būdingus apatinių kvėpavimo takų ligoms, limfmazgių ir tonzilių hipoplazijoms.
Gydymas.
Pakaitinė terapija: į veną leidžiami imunoglobulino preparatai visą gyvenimą skiriami kas 3-4 savaites. Imunoglobulinų dozės parenkamos taip, kad paciento serume susidarytų koncentracija, sutampanti su apatine amžiaus normos riba.
Aptarkite genų terapijos galimybę - geną Btkklonuotas, tačiau jo ekspresija siejama su piktybine kraujodaros audinio transformacija.
Nuolatinės neutropenijos atveju naudojami augimo faktoriai. Kai atsiranda autoimuninės patologijos požymių, galima skirti monokloninius antikūnus (infliksimabą ir kt.).
Dažnas kintamasis imuninis trūkumas
Bendras kintamasis imuninis deficitas (CVID) yra sindromų grupė, kuriai būdingas antikūnų sintezės ir ląstelių imuniteto defektas. Patikimas CVID diagnostinis kriterijus yra reikšmingas dviejų ar trijų pagrindinių izotipų imunoglobulinų kiekio sumažėjimas abiejų lyčių atstovams kartu su vienu iš šių požymių:
Ligos pradžia nuo 2 metų amžiaus;
Izohemagliutininų trūkumas ir (arba) silpnas atsakas į vakcinaciją;
Kitų agammaglobulinemijos priežasčių neįtraukimas.
Kai kuriems pacientams CVID vystymosi priežastis yra genų, koduojančių molekules, dalyvaujančias B ląstelių brendimo ir išgyvenimo procesuose, mutacijos: BAFF-R (B ląstelių aktyvuojančio faktoriaus receptorius),Blimp-1 (B limfocitų sukeltas brendimo baltymas-1)ir ICOS (Indukuojamas kostimuliatorius).Pažeidžiamas B-limfocitų gebėjimas diferencijuotis į plazmos ląsteles, išsivysto antikūnų gamybos defektai, galimi T-limfocitų disfunkcijos, pastebimas padidėjęs polinkis į infekcines ligas. Sindromas gali pasireikšti ankstyvoje vaikystėje, paauglystėje ar jaunuose suaugusiuose.
Laboratorinių tyrimų duomenys.IgG ir IgA (maždaug 50% pacientų) ir IgM (iki nenustatomo kiekio) lygis yra žymiai sumažėjęs. B-limfocitų kiekis kraujyje yra normalus arba sumažėjęs. T-limfocitų skaičius daugumoje pacientų yra normalus. Sunkiems pacientams gali išsivystyti limfopenija (mažiau nei 1500x10 3 ląstelių 1 litre kraujo). NK ląstelių skaičius yra sumažėjęs. Specifinių antikūnų gamyba, reaguojant į imunizaciją, sumažėja arba jo nėra. Labai pakenktas limfocitų dauginimasis ir IL-2 susidarymas veikiant mitogenams ir antigenams.
Klinikinis vaizdas.Aptinkamos pasikartojančios bakterinės infekcinės ligos, lokalizuojančios daugiausia kvėpavimo takus ir paranalinius sinusus. Diagnozės metu kvėpavimo takų infekcijos gali pereiti į bronchektazę ir difuzinius plaučių audinio pažeidimus. Galimas infekcinis virškinimo sistemos pažeidimas, pasireiškiantis viduriavimu, steatorėja ir malabsorbcija (ir, atitinkamai, svorio kritimu). Infekcijos, kurias sukelia Giardia lamblia, Pneumocystis cariniiarba šeimos virusai Herpetoviridae.CVID sergantiems pacientams būdingas pūlingas artritas, kurį sukelia mikoplazmos ir ureaplazmos. Enterovirusinės infekcijos pasireiškimai gali būti encefalomielitas, poliomielitas ir panašūs į dermatomiozitą sindromai, odos ir gleivinės pažeidimai. Autoimuninisligos yra sunkios ir gali nulemti CVID prognozę. Kartais pirmieji klinikiniai CVID pasireiškimai yra artritas, opinis kolitas ir Krono liga, sklerozuojantis cholangitas, malabsorbcija, SLE, nefritas, miozitas, autoimuninė plaučių liga, pasireiškianti limfoidinio intersticinio pneumonito forma, neutropenija,
trombocitopeninė purpura, hemolizinė anemija, žalinga anemija, totalinė alopecija, tinklainės vaskulitas, jautrumas šviesai. Pacientams, sergantiems CVID, į sarkoidozę panašių granulomų ir nepiktybinės limfoproliferacijos dažnis (15% atvejų) žymiai padidėja. Gydymas.
Antibakterinė chemoterapija.
Pakaitinė terapija: į veną leidžiami imunoglobulino preparatai visą gyvenimą skiriami kas 3-4 savaites.
Autoimuninių komplikacijų atveju - imunosupresinis gydymas (gliukokortikoidai, azatioprinas, ciklosporinas A) ir galbūt skiriami monokloniniai antikūnai (infliksimabas ir kt.).
Hyper-IgM sindromai
Hiper-IgM sindromai yra gana retos ligos, kurioms būdingas ryškus IgG, IgA sumažėjimas arba visiškas nebuvimas ir normali ar padidėjusi IgM koncentracija serume. Taip yra dėl B-limfocitų nesugebėjimo perjungti imunoglobulino klases ir kintamų domenų hipermutagenezės. Iki šiol buvo nustatyti 6 genetiniai defektai, dėl kurių išsivystė hiper-IgM sindromas.
. 1 tipas (HIGM 1).X susietas CD40 ligando trūkumas (70% hiper-IgM sindromų atvejų), dėl kurio T ląstelės nesugeba veiksmingai sąveikauti su B limfocitais.
. 2 tipas (HIGM 2).Autosominis recesyvinis, susijęs su AID defekto sukeltu citidino deaminazės (geno 1) aktyvavimu Aicda, 12p13)- fermentas, dalyvaujantis keičiant imunoglobulinų klases ir hipermutagenezę.
. 3 tipas (HIGM 3).Autosominis recesyvinis, susijęs su CD40 molekulės geno mutacija. Tuo pačiu metu B ląstelės pačios nesugeba veiksmingai sąveikauti su T limfocitais. Fenotipinės apraiškos yra panašios į 1 tipo.
. 4 tipas (4 HIGM).Autosominis recesyvinis; kai kuriais atvejais atsiranda mutacijos de novo.Susijęs su UNG defektu - uracilo-DNR glikozilaze - fermentu, kuris taip pat dalyvauja
keičiant imunoglobulinų klases, tačiau po AID veikimo. Šiuo atveju hipermutagenezė neturi įtakos ir sindromas vyksta mažiau sunkiai.
. 5 tipas (HIGM 5).Tik klasės keitimo defektas, hipermutagenezė neturi įtakos. Priežastinė mutacija dar nebuvo nustatyta, tačiau, matyt, yra fermento, veikiančio po to, defektas
PAGALBA.
. 6 tipas (HIGM-ED).X susietą, susietą su disidrotine ektodermine displazija, sukelia NEMO (NF-kB moduliatoriaus) trūkumas, dėl kurio sutrinka CD40 signalizacija.
X susietas hiper-IgM sindromasnustatomi dažniau nei kiti. Jis vystosi su CD40L koduojančio geno defektu (CD154, genas yra ant Xq26-q27.2)yra CD40 ligandas. Nepakankamas T-limfocitų CD40L ekspresija lemia tai, kad B-limfocituose esančių imunoglobulinų klases negalima pakeisti IgM nuo kitų izotipų, taip pat pažeidžiamos atminties B-ląstelės, T-ląstelių repertuaras ir Th1-ląstelių atsakas, nukreiptas prieš intraląstelinius mikroorganizmus. Berniukai serga
Laboratorinių tyrimų duomenys.IgG, IgA, IgE nustatyti negalima arba jie nustatomi labai mažais kiekiais. IgM lygis yra normalus (50% atvejų) arba padidėjęs, dažnai reikšmingai. T ir B ląstelių skaičius yra normalus; sumažėjęs T ląstelių proliferacinis atsakas, kurį sukelia antigenai. IgM yra polikloniniai, kartais monokloniniai. Atskleiskite IgM izotipo autoantikūnus (antieritrocitus, antitrombocitus, antitiroidus, antikūnus lygiųjų raumenų audinių antigenams). Limfoidiniame audinyje nėra gemalo centrų, tačiau yra plazmos ląstelių.
Klinikinis vaizdas.Pirmosios apraiškos pasireiškia kūdikystėje ir ankstyvoje vaikystėje. Pakartota infekcijosskirtinga lokalizacija (pirmiausia kvėpavimo takai), įskaitant oportunistinę (sukelta Pneumocystis carinii).Taip pat būdingi virusų pažeidimai (citomegalovirusas ir adenovirusai), Criptococcus neoformans,mikoplazmos ir mikobakterijos. Kriptosporidinė infekcija gali sukelti ūminį ir lėtinį viduriavimą (išsivystantį 50% pacientų) ir sklerozuojantį cholangitą. Anemija, neutropenija, burnos gleivinės išopėjimas, gingivitas, opinis
stemplės pažeidimai, įvairios žarnyno dalys, opinis kolitas. Jie atskleidžia polinkį autoimuniniai sutrikimai(seronegatyvus artritas, glomerulonefritas ir kt.) ir piktybiniai navikai (daugiausia limfoidinis audinys, kepenys ir tulžies takai). Gali išsivystyti limfadenopatija, hepato- ir splenomegalija. Gydymas
Reguliarus pakaitinis gydymas intraveniniu imunoglobulinu.
Antibakterinė chemoterapija. Pneumocistinės pneumonijos profilaktikai ir gydymui naudojami kotrimoksazolas [sulfametoksazolas + trimetoprimas] ir pentamidinas.
Norėdami išvengti kepenų ir tulžies takų pažeidimo, naudokite tik virtą arba filtruotą vandenį, reguliariai atlikite tyrimus (ultragarsu, kepenų biopsija, jei nurodyta).
Gydant neutropeniją ir burnos išopėjimą, naudojami gliukokortikoidai ir granulocitų kolonijas stimuliuojančio faktoriaus preparatai.
Išsivysčius autoimuninėms komplikacijoms, skiriama imunosupresinė terapija (gliukokortikoidai, azatioprinas, ciklosporinas A), taip pat vaistai, pagrįsti monokloniniais antikūnais.
Optimalus gydymas yra kaulų čiulpų transplantacija iš HLA suderinamų donorų (išgyvenamumas 68%, geriausia tai padaryti iki 8 metų).
KOMBINUOTI IMUNODEFICENCIJOS SU PREVENCINIU T-LIMFOCITŲ DEFEKTU
Sunkus kombinuotas imuninės sistemos nepakankamumas
SCID (SCID - Sunkus kombinuotas imuninės sistemos nepakankamumas)- sindromų grupė, kuriai būdingas T-limfocitų lygio sumažėjimas arba visiškas jų nebuvimas ir adaptacinio imuniteto pažeidimas. ... Retikulinė disgenezė, kuriai būdingas pažeistas limfoidinių ir mieloidinių pirmtakų brendimas ankstyvosiose stadijose: neutropenija ir T - B - NK -.
. X susieta SCID, kuri išsivysto dėl genų mutacijos IL-2RG[(CD132, iš viso prie- IL-2, IL-4, IL-7, IL-9, IL-15 ir IL-21 receptorių grandinė), Xq13.1-q21.1,אּ ], o tai lemia receptorių blokavimą ir tikslinių ląstelių nesugebėjimą reaguoti į atitinkamų interleukinų poveikį (daugiau kaip 50% visų SCID atvejų); T - B + NK -.
. Janus3 tirozinkinazės trūkumas [gen JAK3 (19p13.1),ρ ]; su genų defektais - aktyvacijos signalo perdavimas iš bendro priegrandinės IL-2, IL-4, IL-7, IL-9, IL-15, IL-21 į ląstelės branduolį, o tai lemia T- ir NK-ląstelių diferenciacijos sutrikimą; T - B + NK -.
. Baltymų tirozino fosfatazės trūkumas (CD45, genas PTPRC, 1q31-q32);esant geno defektui, Csk kinazės slopinamasis aktyvumas baltymų tirozino kinazei Src sustiprėja dėl sutrikusio TCR ir BCR ITAM domenų fosforilinimo; T - B + NK +.
. Visiškas RAG1 ir RAG2 fermentų trūkumas, kuris suaktyvina imunoglobulinų ir TCR V (D) J segmentų rekombinaciją [genai RAG1ir RAG2 (11p13),ρ ]; T - B - NK +.
. Omenno sindromas (neišsamus RAG1 ir
RAG2) [genai RAG1ir (arba) RAG2 (11p13-p12),r].Ačiū
dėl mažo šių fermentų liekamojo aktyvumo vis tiek išsivysto daugybė T-limfocitų klonų, būdingų odos ir virškinamojo trakto epitelinių audinių antigenams, kur jie dauginasi ir gamina didelį kiekį IL-4 ir IL-5, sukeldami hipereozinofiliją ir likučių B-limfocitų IgE susidarymą. (nesant kitų imunoglobulinų klasių). Būdinga eritrodermija ir pachiderma su alopecija galvos odoje ir antakiuose, sekinančiu viduriavimu, gyvybei pavojingu infekciniu sindromu; hepatosplenomegalija ir limfmazgių hiperplazija.
. SCID padidėjęs jautrumas jonizuojančiajai spinduliuotei. Artemidės branduolinio baltymo defektas [genas DCLRE1C, (10p),r],įtrauktas į fermentų, reikalingų DNR atkūrimui, kompleksą (dalyvauja sujungiant dvigubas grandines), su geno mutacija įvyksta V (D) J-rekombinacijos pažeidimas; T - B - NK +.
. IL-2 trūkumas [genas IL-2, 4q26-q27].
IL-2 receptorių a-grandinės geno (CD25) mutacijos (10p15-p14);T - B + NK +.
IL-7 receptorių a-grandinės geno (CD127) mutacijos (5p13);T - B + NK +.
TAP trūkumas (Antigeno pristatymo vežėjas),reikalingi antigeniniams peptidams pernešti į IL-7 receptoriaus (CD127) geno a grandinės endoplazminį tinklą. (5p13);T - B + NK +.
CD3 grandinės genų (CD3γ, CDδ ir CDε) mutacijos, dėl kurių sumažėja subrendusių T-limfocitų skaičius, sutrinka jų diferenciacija; T - B + NK +.
Baltymų tirozinkinazės ZAP-70 trūkumas [genas ZAP-70 (2q12),r]. Esant genų mutacijai, kenčia TCR ζ grandinės ITAM domenai ir NK ląstelių ITAM turinčių receptorių fosforilinimas, išsivysto selektyvus CD8 + T ląstelių trūkumas (CD4 + T limfocitų kiekis yra normalus, tačiau funkciniai sutrikimai išreiškiami IL 2 ir platinimas).
Adenozino deaminazės trūkumas [genas ada (20q12-q13.11 , p)], dėl ko ląstelėse kaupiasi metabolitai (deoksiadenozino trifosfatas ir S-adenozilo homocisteinas), slopinantys T- ir B-limfocitų dauginimąsi (aprašomi variantai, kurių liga prasideda vėlai); T - B - NK -.
Purino nukleozidų fosforilazės trūkumas [genas pnp (14q11.2),p], todėl ląstelėse kaupiasi deoksiguanozino trifosfatas, kuris slopina T-limfocitų dauginimąsi (gretutiniai sindromai - urikemija ir urikurija); T - B + NK -.
Laboratorinių tyrimų duomenys.Atskleisti kintamą, kartais gilią limfopeniją; limfocitai negali daugintis, reaguodami į specifinį antigeną; dažnai išreiškiamas imunoglobulinų kiekio sumažėjimas kraujo serume. Krūtinės ląstos rentgenogramoje nėra užkrūčio liaukos šešėlio.
Klinikinis vaizdas.Paprastai klinikinė diagnozė paaiškėja per pirmuosius 6 gyvenimo mėnesius, kai motinos IgG antikūnai išnyksta. Klinikiniame vaizde išryškinkite sunkus infekcinis sindromas,limfoidinio audinio hipoplazija ir vystymosi sulėtėjimas. Infekciniam sindromui būdinga burnos ertmės kandidozė, lėtinis viduriavimas, plaučių uždegimas, karščiavimas,
bakterinės etiologijos sepsis, virusinės infekcijos. Infekcijų sukėlėjai priklauso skirtingoms taksonominėms grupėms: bakterijoms, virusams, grybams, oportunistiniams mikroorganizmams (Pneumocystis carinii).Pneumoniją dažnai sukelia P. carinii,viduriavimas - rotavirusai, Kampilobakterija, Giardia lamblia.Virusinis hepatitas dažnai pasireiškia. Būdingas regioninio arba generalizuoto BCGito išsivystymas po vakcinacijos.
Gydymasnumato skirti palaikomąją terapiją, įskaitant parenteralinę mitybą, į veną įvedamo imunoglobulino įvedimą, antibiotikų, priešgrybelinių ir antivirusinių vaistų paskyrimą. Vienas pagrindinių gydymo būdų norint pasveikti yra kaulų čiulpų transplantacija, be kurios vaikai, sergantys SCID, paprastai miršta pirmaisiais gyvenimo metais. Apibūdinami pavieniai atvejai, kai vaikas ypač sanitarinėmis sąlygomis gyveno iki 2–3 metų. Svarbu kuo anksčiau atpažinti naujagimių SCID, nes, pavyzdžiui, jiems imunizacija gyvomis vakcinomis yra mirtina. Iš karto po diagnozės tokius vaikus reikia paguldyti į gnotobiologines sąlygas (sterili dėžutė). Pridedant infekcines ligas, atliekama intensyvi antibakterinė, antivirusinė ir priešgrybelinė terapija, pakaitinė terapija į veną leidžiamu imunoglobulinu. Pneumocistinės pneumonijos profilaktikai skiriamas kotrimoksazolas. BCGitis vystymosi atveju būtina atlikti ilgalaikę intensyvią prieštuberkuliozinę terapiją. Kraujo komponentams perpylti reikia naudoti tik švitintus ir filtruotus vaistus. Dėl motinos limfocitų perkėlimo į transplacentinę riziką yra transplantato prieš šeimininką reakcija.
Nuogų limfocitų sindromas
Tai yra patologijos pavadinimas, kai organizmas neišreiškia MHC-I ar MHC-II molekulių. Nesant MHC-I molekulių ekspresijos, sumažėja CD8 + T-limfocitų kiekis ir nėra NK-ląstelių aktyvumo; nesant MHC-II, CD4 + T-limfocitų lygis yra sumažėjęs. Buvo apibūdinti keli genetiniai defektai. Tačiau šie defektai nėra lokalizuoti MHC genuose, bet keliuose skirtinguose veiksniuose, atsakinguose už jų reguliavimą
išraiška. Klinikinis vaizdas„nuogų“ limfocitų ir gydymasyra panašūs į kitų SCID.
DiGeorge sindromas
Su DiGeorge sindromu arba trečios ir ketvirtos ryklės kišenių sindromu [išbraukta 22q11,įskaitant geną TBX1 (22q11.2),atskleisti užkrūčio liaukos hipoplaziją ar aplaziją, prieskydinės liaukos hipoplaziją, širdies ydas, T-limfocitų trūkumą, kintantį B-limfocitų skaičių.
Laboratorinių tyrimų duomenys.Reikšmingas CD3 +, CD4 + ir CD8 + T ląstelių skaičiaus sumažėjimas ir staigus jų proliferacinio aktyvumo sumažėjimas, kurį sukelia mitogenai ir antigenai. B ir NK ląstelių skaičius yra normalus. Imunoglobulinų koncentracija serume daugeliu atvejų yra normos ribose, galimi įvairūs disgammaglobulinemijos variantai.
Klinikinis vaizdas.Imunodeficito komponentas yra hipoplazija arba užkrūčio liaukos aplazija ir pasikartojanti, sunki užkrečiamos ligos.Taip pat nustatomas hipoparatiroidizmas (hipokalcemija ir dėl to tetanija, pastebima 1-2 dienas po gimimo); kraujotakos sistemos defektai (dešinysis aortos lanko posūkis, dešiniojo skilvelio stenozė, tarpskilvelinės ir tarpatrialinės pertvaros, Fallot tetrados, plaučių arterijos atrezijos ar hipoplazijos defektai); gomurio tuštuma; veido griaučių anomalijos (padidėjęs atstumas tarp suporuotų organų, sumažėję žandikauliai, ypač apatiniai, žemai įsitaisę ausies kaušeliai, trumpas nosies griovelis). Išreikšti gerklų, ryklės, trachėjos, vidinės ausies, stemplės struktūros anomalijos; sutrikusi inkstų, centrinės nervų sistemos ir kitų apsigimimų raida (polidaktilija, nagų nebuvimas, išangės atrezija, išangės fistulės). Būdingas kalbos vėlavimas ir psichomotorinis vystymasis. Jie atkreipia dėmesį į polinkį autoimuniniai sutrikimai(citopenija, autoimuninis tiroiditas) ir piktybiniai navikai.
Gydymas.... Antibakterinis ir antivirusinis gydymas. ... Pakaitinė terapija į veną leidžiamais imunoglobulino preparatais. ... Chirurginis gydymas, siekiant ištaisyti apsigimimus. ... Dėl autoimuninių komplikacijų - imunosupresinė terapija. ... Esant endokrinopatijoms, atitinkamų sutrikimų korekcija. ... Kaulų čiulpų transplantacija yra neveiksminga
tivna. ... Tyminio epitelio audinio transplantacija yra pagrįsta. Parathormoninių liaukų funkcijos korekcija.
X susietas limfoproliferacinis sindromas
X- susietam limfoproliferaciniam sindromui būdingas sutrikęs imuninis atsakas į Epstein-Barr virusą [kurį sukelia genų defektai SH2D1A(SAP) „Xq25“,אּ], dėl to nekontroliuojami B-limfocitų, transformuotų su Epstein-Barr virusu, dauginimasis ir virusas užkrėtė naujas tikslines ląsteles.
Klinikinis vaizdas.Apibūdinami 4 dažniausiai pasitaikantys fenotipai: sunki infekcinė mononukleozė, piktybinės limfoproliferacinės būklės (limfomos, leukemijos, daugiausia B ląstelės), anemija arba pancitopenija (įskaitant tuos, kuriuos sukelia viruso sukeltas hemofagocitinis sindromas), disgammaglobulinemija. Infekcija Epstein-Barr virusu yra sunkiausių, greitai progresuojančių ir mirtinų ligų susidarymo (paleidimo) mechanizmas: fulminantinė infekcinė mononukleozė (mirtina 58% atvejų), hemofagocitinis sindromas (be gydymo, 100% atvejų - mirtinas). 10% atvejų fenotipas pasireiškia prieš užsikrėtus Epstein-Barr virusu (dažniausiai išsivysto disgammaglobulinemija ir limfomos). Dažniausiai nustatomos įvairios hipogammaglobulinemijos rūšys. Dėl imunodeficito atsiranda bakterijų, grybelių ir virusų užkrečiamos ligos.Įtarti ligą galima berniukams, turintiems būdingą šeimos istoriją ir serologinį arba PGR teigiamą Epstein-Barr viruso testą. Diagnozei rekomenduojama naudoti genetinės analizės derinį SH2D1Air vertinant SAP išraiškos lygį.
Gydymas
Profilaktikos tikslais rekomenduojama vartoti antivirusinius vaistus - aciklovirą, valaciklovirą (jų ankstyvas vartojimas slopina Epstein-Barr viruso replikaciją burnos ertmėje) ir į veną leidžiamą imunoglobuliną (su dideliu antikūnų titru Epstein-Barr virusui).
Sergant hipogammaglobulinemija, į veną leidžiamas imunoglobulinas vartojamas kas mėnesį kartu su antibiotikų terapija.
Fulminant infekcinei mononukleozei gydyti skiriamos didelės acikloviro ir metilprednizolono dozės, skiriamos didelės dozės į veną leidžiamu imunoglobulinu su dideliu antikūnų prieš Epstein-Barr virusą ir IFNa titru.
Išsivysčius hemofagocitiniam sindromui, didelės deksametazono dozės derinamos su vepezidu ♠ (etopozidu).
Gydant piktybines ligas, naudojami standartiniai terapijos protokolai.
Radikalus gydymas yra kaulų čiulpų transplantacija iš HLA suderintų donorų.
Autoimuninis limfoproliferacinis sindromas
Autoimuninis limfoproliferacinis sindromas yra ligų grupė, kuriai būdinga gerybinė limfoproliferacija, hiperimunoglobulinemija, autoimuniniai sutrikimai, padidėjęs CD3 + CD4 - CD8 - T-limfocitų (dvigubai neigiamas) kiekis periferiniame kraujyje ir apoptozės defektas [geno defektas] Fas(CD95) - TNFRSF6 (10q24,1),genas kaspazė-10,Faso ligandas - FasL (1q23)].
Laboratorinių tyrimų duomenys.CD3 + CD4 - CD8 - T-limfocitų kiekis periferiniame kraujyje ar limfoidiniuose audiniuose yra didesnis nei 1%. IgG, IgA ir IgM kiekis gali būti normalus, padidėjęs ar net sumažėjęs. Su amžiumi hipergammaglobulinemiją pakeičia maža imunoglobulinų koncentracija serume, iki agammaglobulinemijos. Atskleisti eritrocitų, trombocitų, neutrofilų, lygiųjų raumenų ir VIII faktoriaus autoantikūnus; antinukleariniai ir antifosfolipidiniai autoantikūnai, taip pat reumatoidinis faktorius ir kt. Būdinga limfocitozė.
Klinikinis vaizdas.Visiems pacientams yra padidėjusios kepenys, limfmazgiai (per pirmuosius 5 gyvenimo metus) ir blužnis. Limfoproliferacija nėra lydima karščiavimo ir naktinio prakaitavimo. Debiutas autoimuninės reakcijosgali nesutapti su limfoproliferacija ir įvykti vėliau. Su amžiumi autoimuninių reakcijų sunkumas didėja. Dažniau išsivysto autoimuninės reakcijos prieš kraujo ląsteles (hemolizinė anemija, trombocitopenija, neutropenija), rečiau pažeidžiami kiti organai. Padidėja piktybinių navikų išsivystymo rizika (T ir B limfomos, Burkitto limfoma, netipinė limfoma, limfogranulomatozė ir kt.).
Gydymas.... Chemoterapiniai vaistai (ciklofosfamidas, azatioprinas, metotreksatas, chlorambucilas). ... Gliukokortikoidai. ... Splenektomija su sunkiu hipersplenizmu ir hemocitopenija. ... Sunkiais atvejais galima kaulų čiulpų transplantacija.
Hiperimunoglobulinemijos E sindromas
Hiper-IgE sindromui būdingas reikšmingas IgE lygio padidėjimas serume, pasikartojantys stafilokokų etiologijos odos ir poodinio audinio abscesai, pneumonija, susidarant pneumocelei, anomalijos veido griaučių struktūroje, AD. Hiper-IgE sindromo molekulinė genetinė prigimtis dar nėra nustatyta. Vienais atvejais buvo atskleistas autosominis dominantas, kitais - autosominis recesyvinis paveldėjimas. Daroma prielaida, kad defektai turi įtakos signalinėms citokinų receptorių molekulėms (šio sindromo autosominėje dominuojančioje formoje, Stat3)ir, galbūt, yra susiję su Th17 subpopuliacijos disfunkcija. Kitas genas, atsakingas už hiper-IgE sindromo susidarymą, yra lokalizuotas 4 chromosomoje (4q).
Laboratorinių tyrimų duomenys.Aptinkami įvairūs imunologiniai sutrikimai: padidėjęs IgE kiekis serume, neutrofilų chemotaksės pažeidimas, antikūnų susidarymo defektas; PHT atsako į kandidiną, difteriją ir stabligės toksoidą sumažėjimas; susilpnėjęs T ląstelių proliferacinis aktyvumas reaguojant į Candidair stabligės toksoidas, išlaikant atsaką į mitogenus. Eozinofilija periferiniame kraujyje ir odos abscesų skystyje. T ir B ląstelių skaičius yra normalus.
Klinikinis vaizdas.Vidutinė egzema ankstyvame amžiuje. Būdingi veido bruožai (platus nosies tiltelis, plati išsišakojusi nosis, veido griaučių asimetrija, ryški kakta, giliai įsišaknijusi akis, aukštas gomurys). Jie atskleidžia skeleto vystymosi anomalijas, skoliozę, padidėjusį sąnarių judrumą, polinkį į kaulų lūžius po nedidelių sužalojimų, dantų pakeitimo pažeidimą. Yra odos, poodinio audinio ir limfmazgių abscesai. Pneumonija išsivysto vyresniame amžiuje (dažniausiai patogenai S. aureusir H. in-
gripai),77% atvejų susidaro pneumocelė, infekcija dėl P. aeruginosair A. fumigatus.Pneumonija gali vykti be karščiavimo. 83% atvejų išsivysto lėtinė gleivinės ir nagų kandidozė.
Gydymas.... Ilgalaikė (profilaktikos tikslu - visą gyvenimą) antibakterinė ir priešgrybelinė terapija. ... Dermatitui gydyti vietiniai vaistai naudojami, sunkiais atvejais - mažos ciklosporino A dozės. Kaulų čiulpų transplantacija yra neveiksminga.
CHROMOSOMŲ GEDIMO SINDROMAI
Sindromams su chromosomų nestabilumu: ataksiateleangiektazija[DNR topoizomerazės geno defektas Bankomatas (11q22),p] ir nijmegeno sindromas[Nibrino geno defektas NBS1(8q21)] - būdingas padidėjęs piktybinių navikų dažnis, savaiminis chromosomų nestabilumas ir chromosomų skaidymas. Abu baltymai dalyvauja DNR dvigubų grandžių pertraukų taisyme ir ląstelių ciklo reguliavime. Paprastai dvigubos grandinės DNR pertraukos įvyksta per V (D) J rekombinaciją imunoglobulino ir TCR genuose, perjungiant imunoglobulino klases, perėjus per mejozę. Panašūs procesai vyksta brandinant neuronus smegenyse. Ataksijos-telangiektazijos ir Nijmegeno sindromo DNR atstatymo defektai sukelia tokias klinikines apraiškas kaip imunoglobulinų sintezės sutrikimai, lytinių organų ir nervų sistemos funkcija.
Ataksija-telangiektazija
Šį labai heterogeninio fenotipo sindromą (dažnis 1: 300 tūkst. Naujagimių) apibūdino prancūzų gydytojas D. Louis-Baras. Ataksijos simptomus galima nustatyti jau 2–4 mėnesių amžiaus vaikui. Ataksiją sukelia laipsniškas Purkinje ląstelių degeneracija smegenėlėse. Nosies, ausų, junginės odos telangiektazijos atsiranda šiek tiek vėliau, praėjus 3-6 metams. Dažnai ant odos atsiranda kavos su pienu dėmių. Būdinga užkrūčio liaukos, limfmazgių, blužnies, tonzilių hipoplazija. Imunodeficitas pasireiškia IgA, IgE, IgG2, IgG4 gamybos sumažėjimu (dažnai disbalansu). Išsivysto 80% pacientų
yra atitinkama infekcinė klinikinė simptomatologija. Sumažėjęs T ląstelių (daugiausia CD4 + T ląstelių) skaičius ir funkcinis aktyvumas. Bendras T limfocitų skaičius daugumoje pacientų yra normalus. Neįprastai didelis (200 kartų didesnis nei bendroje populiacijoje) neoplazmų (daugiausia limfomų ir karcinomų) dažnis, dėl kurio 10–12 metų dažnai miršta. Gydymassimptominis.
Nijmegeno sindromas
Nijmegeno sindromas (po miesto pavadinimo Olandijoje, kur pirmą kartą buvo aprašyta liga) pasireiškia mikrocefalija, specifiniais veido griaučių sutrikimais (nuožulni kaktos, išsikišusios vidurinės veido dalies, ilgos nosies, apatinio žandikaulio hipoplazijos, mongoloidinio akies pjūvio, epikanto, didelių ausų), uždelsto fizinio vystymosi. , dėmių "kava su pienu" buvimas ant odos; klinodaktilija ir sindaktilija, kiaušidžių disgenezė ir kt. Daugumą vaikų kenčia nuo pasikartojančių ir lėtinių bakterijų užkrečiamos ligoskvėpavimo takai, ENT organai ir šlapimo sistema. 50% atvejų išsivysto piktybiniai navikai, daugiausia B ląstelių limfomos. Jie atskleidžia įvairias disgammaglobulinemijos formas, CD4 + T ląstelių sumažėjimą.
Gydymas.... Simptominė neurologinių sutrikimų terapija. ... Pakaitinė terapija į veną leidžiamu imunoglobulinu. ... Pagal indikacijas taikoma antibakterinė, antivirusinė, priešgrybelinė terapija. ... Gydant piktybinius navikus, atsižvelgiama į padidėjusį jautrumą radiacijai ir chemoterapijai.
Wiskott-Aldrich sindromas
Wiskott-Aldrich sindromas [geno defektas WASP (Xp11.23p11.22),אּ; taip pat ρ ir Ʀ] Gen WASP(iš Wiskott-Aldrich sindromas)ekspresuojamas limfocituose, blužnies audinyje ir timocituose. Šio geno mutacijos yra susijusios su nenormalia CD43 molekulės ekspresija neutrofiluose ir T-limfocituose (CD4 ir CD8) (ICAM-1 ligandas turi antiadhesinę funkciją).
Laboratorinių tyrimų duomenys.Trombocitopeniją (mažiau nei 10% normos) sukelia padidėjęs ląstelių sunaikinimas.
Trombocitai yra mažesni nei sveikų žmonių. Serumo IgM lygis sumažėja esant normaliam IgG lygiui ir padidėjus IgA bei IgE. Sumažėję izohemagliutininų titrai, sutriko antikūnų susidarymas prieš pneumokoko, streptokoko, Escherichia coli, Salmonella polisacharidų antigenus, taip pat antivirusinius antikūnus. Ankstyvame amžiuje limfocitų skaičius paprastai yra normalus; po 6 metų nustatoma limfopenija (mažiau nei 1x10 9 / l), sumažėja CD3 + ir CD4 + T ląstelių, kurių B ir NK ląstelių lygis yra normalus. Galima eozinofilija ir posthemoraginės anemijos išsivystymas. Sumažėja T ląstelių proliferacinis atsakas į mitogenus ir antigenus, susilpnėja PHT. Blužnyje normali gemalo centrų ir T ląstelių zonų struktūra nėra aptinkama.
Klinikinis vaizdas.Ligai būdinga simptomų trijulė: trombocitopenija, egzema ir pasikartojančios infekcinės ligos.Hemoraginis sindromas pasireiškia anksti, jau naujagimio laikotarpiu (petechialinis bėrimas, cefalohematomos, kraujavimas iš bambos žaizdos, kraujavimas iš žarnyno). Egzema pasireiškia nuo ankstyvo amžiaus 80% pacientų. Imunodeficito požymiai didėja su amžiumi: bakterinės infekcinės ENT organų, kvėpavimo sistemos, virškinimo organų, odos ligos; paplitusi ar apibendrinta herpeso infekcija (Paprastoji pūslelinėir Varicella zoster),citomegalovirusas, taip pat grybelinė (gleivinės kandidozė), rečiau oportunistinės infekcinės ligos. Autoimuninės ligos (hemolizinė anemija, neutropenija, artritas, odos vaskulitas, opinis kolitas, smegenų vaskulitas, glomerulonefritas, autoimuninė trombocitopenija) nustatomos 70% pacientų. Vyresniems nei 5 metų pacientams dažnis piktybiniai navikai(daugiausia limfoidinio audinio navikai).
Gydymas.... Alogeninių kaulų čiulpų ar kamieninių ląstelių transplantacija (operacijos sėkmės procentas pasiekia 90%, kai naudojamas transplantatas iš histokompatiblaus donoro, o 50% - su haploidentine transplantacija). ... Pakaitinė terapija į veną leidžiamu imunoglobulinu. ... Profilaktinis antibakterinių, priešgrybelinių ir antivirusinių vaistų vartojimas. ... Siekiant sumažinti hemoraginį sindromą, atliekama splenektomija. ... Esant autoimuninėms komplikacijoms, skiriama imunosupresinė terapija.
FAGOCITOZĖS DEFEKTAI
Lėtinė granulomatozinė liga
Lėtinė granulomatozinė liga pasižymi sutrikusiu fagocitų funkciniu aktyvumu (aktyvių deguonies radikalų formų susidarymu, ląstelių žudymu ir fagocitozuotų patogenų fragmentavimu), nuolatinėmis bakterinėmis ir grybelinėmis infekcinėmis ligomis bei granulomatozinio uždegimo išsivystymu. Lėtinė granulomatozinė liga vystosi asmenims, turintiems įvairių genetinių defektų [65% atvejų - su liga susijęs X variantas: genas gp91-phox (Xp21.1),אּ; 35% atvejų - autosominis recesyvinis: genas f47-phox (7q11.23),ρ; genas p67-phox (1q25),ρ; genas p22-phox (16q24),p], dėl ko sutrinka NADP-oksidazės sistema. Mirus trumpalaikiams (kelias valandas) neutrofilams, neužmuštos bakterijos „teka“ į uždegimo židinį. Makrofagai yra ilgaamžės ląstelės, o jų pirmtakai (monocitai) padidėjusiu skaičiumi migruoja į židinį (dėl to susidaro granulomos),fagocitozės mikroorganizmus, tačiau nesugeba jų sunaikinti.
Laboratorinių tyrimų duomenys.Imunoglobulinai serume ir limfocitų pogrupiai yra normalūs. Peroksidų radikalų susidarymas neutrofiluose, įvertintas atliekant bandymus (nuo liuminolio priklausoma chemiliuminescencija arba tetrazolio mėlynos spalvos redukcija), smarkiai sumažėja arba jo nėra. Infekcinių ligų fone būdinga leukocitozė, neutrofilija, padidėjęs ESR, anemija ir hipergammaglobulinemija.
Klinikinis vaizdas.Liga daugeliu atvejų pasireiškia pirmaisiais gyvenimo metais. infekcinis sindromas(infekcijos intra- ir tarpląsteliniuose patogenuose) ir granulomų susidarymas. Būdingiausi: plaučių pažeidimai (pasikartojanti pneumonija, linksmųjų limfmazgių pažeidimai, plaučių abscesai, pūlingas pleuritas), virškinamojo trakto, odos abscesai ir limfadenitas. Dažniausiai patogenai yra teigiami katalazės mikroorganizmai: S. aureus, Aspergillus spp.,žarnyno gramneigiamos bakterijos (E. coli, Salmonella spp., Serratia marcescens),ne taip dažnai - Burkholderia cepaciair Nocardia farcinica.Būdingas kepenų ir subfrenijos abscesų, osteomielito, pararektalinių abscesų, sepsio vystymasis.
Sunkiausia, gyvybei pavojinga infekcinė komplikacija yra aspergiliozė, kuri gali pasireikšti difuzine plaučių ir kitų organų (riebalinio audinio, smegenų, kaulų, sąnarių, endokardo) pažeidimu. Pacientams, sergantiems lėtine granulomatine liga po vakcinacijos BCG, dažnai išsivysto su vakcina susijusi infekcija, apimanti regioninius limfmazgius. Mikobakterijų pažeidimai pacientams, sergantiems lėtine granulomatozine liga, gali lokalizuotis tiek plaučiuose, tiek ekstrapulmonaliai ir užsitęsti. Pacientams, sergantiems lėtine granulomatozine liga, būdingas fizinio išsivystymo atsilikimas.
Gydymas.... Antimikrobinė terapija: profilaktinis nuolatinis kotrimoksazolo ir priešgrybelinių vaistų (itrakonazolo ir kt.) Vartojimas; esant infekcinėms komplikacijoms, kombinuotas gydymas antibiotikais (2-3 baktericidiniai antibiotikai, prasiskverbiantys į ląstelę) kartu su priešgrybeliniu gydymu atliekamas parenteraliai. Išsivysčius aspergiliozei, reikia ilgalaikio amfotericino B ar kaspofungino vartojimo. Sergant mikobakterine infekcija, naudojamas ilgalaikis specifinis gydymas kartu su vaistais nuo tuberkuliozės ir plataus spektro antibiotikais. ... Chirurginis gydymas dažnai lydimas pooperacinės žaizdos supūtimo ir naujų židinių susidarymo. Galimas punkcinis absceso nutekėjimas kontroliuojant ultragarsu. ... Gydant sunkias infekcines komplikacijas, kai antibiotikų terapija yra neveiksminga, galima naudoti granulocitų masę, dideles IFNu ir G-CSF dozes. ... Suderinto brolio ir kaulų čiulpų transplantacija arba virkštelės kraujo ląstelių transplantacija gali būti sėkminga ankstyvame amžiuje, kai mirties nuo infekcinių komplikacijų ir transplantato prieš šeimininką ligos rizika yra minimali.
Leukocitų adhezijos defektai
Iki šiol buvo aprašyti 3 leukocitų adhezijos defektai. Jie visi turi
autosominis recesyvinis paveldėjimas, kuriam būdingos pasikartojančios ir lėtinės bakterinės ir grybelinės infekcinės ligos. I tipui būdingas CD11 / CD18 ekspresijos nebuvimas ar sumažėjimas leukocituose, sutrikusi neutrofilų chemotaksija, leukocitozė (daugiau nei 25x10 9), vėlyvas virkštelės netekimas ir omfalito išsivystymas, prastas žaizdų gijimas, pūlių susidarymo stoka patogeno įsiskverbimo į kūną vietoje.
Gydymas.... Antibiotikų terapija: infekciniai epizodai ir profilaktika. ... Sunkiais atvejais skiriama kaulų čiulpų transplantacija iš HLA suderinamo donoro.
RINKINYS SISTEMOS DEFEKTAI
Ligos, kai trūksta komplemento komponentų
Atskirų komplemento sistemos komponentų genų defektų apraiškos parodytos lentelėje. 11–2.
Paveldima UAB.Ligos, kurias sukelia komplemento komponentų trūkumas, nustatomos retai, nes jų pasireiškimui būtina autosominių alelių homozigotinė būsena. Yra viena išimtis, susijusi su C1inh (C1 esterazės inhibitorius): geno mutacija C1inh,sukeliantis inhibitoriaus trūkumą, heterozigotinėje būsenoje pasireiškia fenotipu, žinomu kaip paveldimas AO (daugiau informacijos žr. 13 skyrių „Angioedema“).
Imuninių kompleksų ligos.C1-C4 nepakankamumas pasireiškia išsivysčius imuninių kompleksų ligoms - sisteminiam vaskulitui ir inkstų pažeidimams, kurie bendrai vadinami sisteminės raudonosios vilkligės (SLE) sindromu.
Pirogeninės infekcijos.C3 trūkumas (taip pat H ir I faktoriai) yra susijęs su padidėjusiu jautrumu pirogeninėms infekcijoms. Komponentų, dalyvaujančių alternatyviame komplemento aktyvacijos kelyje, trūkumai, taip pat C5-C8 komponentų trūkumai yra susiję su padidėjusiu jautrumu infekcijai, kurią sukelia Neisseria spp.C9 trūkumas paprastai būna kliniškai besimptomis.
11-2 lentelė.Atskirų komplemento sistemos komponentų defektų klinikiniai pasireiškimai
Manozę surišančio lektino trūkumas
Manozę surišančio lektino (taip pat žinomo kaip manozę surišančio lektino - MSL) trūkumas yra dėl geno defekto MBL(įvairios geno taškinės mutacijos ir ištrynimai MBLnustatyta 17% Kaukazo rasės žmonių). Esant genų defektams, sutrinka proteazių, skaidančių komplemento komponentus C2 ir C4, aktyvavimas ir komplemento sistemos aktyvavimas palei lektino kelią. Kliniškaiši patologija pasireiškia infekciniu sindromu.
Laboratorinių tyrimų duomenys.Analizuojant limfocitų, leukocitų, taip pat imunoglobulino izotipų pogrupius, reikšmingų pakitimų, adekvačių klinikiniams simptomams, nėra. Kraujo serume nėra MSL.
Gydymas.Ši liga nėra klasikinis imunodeficito sutrikimas. Todėl imunokorekcija imunotropiniais vaistais yra draudžiama. Rekombinantinė MSL gali būti naudojama kaip farmakologinė priemonė etiopatogenezinei pakaitinei terapijai pacientams, turintiems šį paveldimą defektą. Šiuo metu šiuo vaistu atliekami klinikiniai tyrimai.
Vaikų laikina hipogammaglobulinemija
Vaikų laikina hipogammaglobulinemija yra susijusi su fiziologiniu imunoglobulino sistemos fazinio formavimosi ypatumu. Labiausiai „vėluoja“ IgM ir IgA antikūnų susidarymas. Sveikiems vaikams motinos IgG kiekis palaipsniui mažėja, o po šešių mėnesių padidėja jų pačių IgG antikūnų gamyba. Tačiau kai kurių vaikų imunoglobulino koncentracijos padidėjimas vėluoja. Tokie vaikai gali sirgti pasikartojančiomis bakterinėmis infekcinėmis ligomis. Tokiais atvejais donoro imunoglobulino infuzijos (į veną leidžiamo imunoglobulino) vartoti negalima.
Selektyvus imunoglobulino A trūkumas
Selektyvus imunoglobulino A (SD IgA - Selektyvus IgA trūkumas)išsivysto dėl genų defekto tnfrsf13b
arba p). IgA trūkumas, esant kitų klasių imunoglobulinams, yra dažniausias imunodeficitas, nustatytas visoje populiacijoje, kurio dažnis yra 1: 500-1500 žmonių (dar dažniau alergiškiems pacientams). Atskirkite selektyvų IgA trūkumą, t.y. susideda iš vieno poklasio trūkumo (30% atvejų) ir visiško (70% atvejų). IgA2 poklasio trūkumas lemia ryškesnį klinikinį vaizdą nei IgA1 poklasio trūkumas. Taip pat galimi IgA trūkumo deriniai su kitais sutrikimais: su IgG biosintezės defektu ir su T-limfocitų anomalijomis. Didžioji dauguma žmonių, turinčių selektyvų
igA trūkumas yra praktiškai sveikas. Vaikams iki 2 metų IgA trūkumas yra fiziologinė būklė.
IgA koncentracijos serume sumažėjimas iki<5 мг/дл у детей старше 4 лет; IgG и IgM в норме, количество и соотношение субпопуляций лимфоцитов и их функциональная активность могут быть в норме.
Klinikinis vaizdas.Esant IgA trūkumui, gali išsivystyti 3 patologinių sindromų grupės: infekciniai, autoimuniniai ir alerginiai. Pacientai, turintys IgA trūkumą, yra linkę pasikartoti viršutinių kvėpavimo takų ir virškinimo sistemos infekcijose. Dažniausios ir sunkiausios yra įvairios autoimuninės ligos (reumatoidinis artritas, ankilozuojantis spondilitas, Sjogreno sindromas, vaskulitas su smegenų kraujagyslių pažeidimu, autoimuninis tiroiditas, SLE, glomerulonefritas, hemolizinė anemija, I tipo cukrinis diabetas, vitiligo ir kt.) Celiakijos dažnis yra 10 kartų didesnis nei vaikams, kurių IgA yra normali. Dažniausiai nustatomos alerginės apraiškos: karvės pieno baltymų netoleravimas, atopinis dermatitas (AD), bronchinė astma.
Gydymas.Asimptominiams atvejams nereikia specialaus gydymo; esant infekcinių, autoimuninių ir alerginių ligų klinikinėms apraiškoms, gydymas atliekamas pagal standartus.
Pakaitinė terapija donoriniais imunoglobulinais nenurodyta nei esant selektyviam, nei visiškam IgA trūkumui, nes yra didelė tikimybė, kad recipiente susidarys anti-izotipiniai antikūnai prieš IgA ir atsiras jų sukeltų perpylimo komplikacijų.
Agammaglobulinemija su B ląstelių trūkumu
Su X susijusi agammaglobulinemija (Brutono liga)sudaro 90% visų agammaglobulinemijos atvejų. Serga defektinio geno nešiotojai, berniukai, sūnūs (אּ, ρ) bTK (Xq21.3-q22),koduojantis B limfocitams būdingą baltymų tirozino kinazę Btk (Brutono tirozino kinazė- Brutono tirozino kinazė). Dėl defekto pažeidžiami tarpląsteliniai signalizacijos keliai, sunkiųjų imunoglobulinų grandinių rekombinacija,
pre-B ląstelių perteikimas B-limfocitams. 10% pacientų agammaglobulinemija su B ląstelių trūkumu paveldima autosominiu recesyviniu būdu. Šiuo metu aprašyti 6 genetiniai defektai, įskaitant prieš B ląstelių receptorių, citoplazminį B ląstelių adapterio baltymą (BLNK) ir geną Kartotinai turintis leucino, kuriame yra 8 (LRRC8).
Laboratorinių tyrimų duomenys.Periferinių B limfocitų nėra. Kaulų čiulpuose yra prieš B ląstelių, kurių citoplazmoje yra μ grandinė. T-limfocitų skaičius ir T-limfocitų funkcijos tyrimai gali būti normalūs. IgM ir IgA kraujyje nustatyti negalima; IgG gali būti, tačiau mažais kiekiais (0,4–1,0 g / l). Antikūnų prieš kraujo grupių antigenus ir vakcinos antigenus (stabligės, difterijos toksinų ir kt.) Nėra. Gali išsivystyti neutropenija. Histologinis limfoidinio audinio tyrimas: limfoidiniuose folikuluose nėra gemalo (embriono) centrų ir plazmos ląstelių.
Klinikinis vaizdas.Jei nežinoma šeimos istorija, diagnozė paaiškėja vidutiniškai 3,5 metų. Ligai būdinga limfoidinio audinio hipoplazija, sunkios pūlingos infekcijos, viršutinių (sinusito, vidurinės ausies uždegimo) ir apatinių (bronchito, plaučių uždegimo) kvėpavimo takų infekcinės ligos; galimas gastroenteritas, pioderma, septinis artritas (bakterinis ar chlamidinis), septicemija, meningitas, encefalitas, osteomielitas. Kvėpavimo ligų sukėlėjai dažniausiai yra Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus,viduriavimas žarnyno bakterijos arba lamblia Giardia lamblia.Taip pat pacientai, sergantys agammaglobulinemija, yra linkę į mikoplazmų ir ureaplazmų sukeltas infekcines ligas, kurios yra lėtinės pneumonijos, pūlingo artrito, cistito ir poodinio audinio abscesų atsiradimo priežastis. Tipiški virusai yra neurotropiniai virusai ECHO-19 ir coxsackie, kurie sukelia ir sunkų ūminį, ir lėtinį encefalitą, ir encefalomielitą. Enterovirusinių infekcijų apraiška gali būti į dermatomiozitą panašus sindromas, ataksija, galvos skausmai ir elgesio sutrikimai. Sergantiems vaikams, skiepijant gyva poliomielito vakcina, paprastai nustatomas ilgalaikis poliomielito viruso išsiskyrimas per gleivinę ir atkurtas bei didėjantis virulentiškumas (t. Y.
yra realus sveikų vaikų, užsikrėtusių poliomielitu, užkrėtimo pavojus dėl kontakto su vakcinuotu imunodeficito vaiku). Autoimuniniai sutrikimai sergant agammaglobulinemija gali pasireikšti reumatoidiniu artritu, į sklerodermą panašiu sindromu, skleredema, opiniu kolitu, I tipo cukriniu diabetu (dėl vyraujančio Th1 imuninio atsako).
Medicininė apžiūra.Atkreipkite dėmesį į fizinio vystymosi atsilikimą, į pirštų formą (pirštai būgnelių pavidalu), krūtinės formos pokyčius, būdingus apatinių kvėpavimo takų ligoms, limfmazgių ir tonzilių hipoplazijoms.
Gydymas.
Pakaitinė terapija: į veną leidžiami imunoglobulino preparatai visą gyvenimą skiriami kas 3-4 savaites. Imunoglobulinų dozės parenkamos taip, kad paciento serume susidarytų koncentracija, sutampanti su apatine amžiaus normos riba.
Aptarkite genų terapijos galimybę - geną Btkklonuotas, tačiau jo ekspresija siejama su piktybine kraujodaros audinio transformacija.
Nuolatinės neutropenijos atveju naudojami augimo faktoriai. Kai atsiranda autoimuninės patologijos požymių, galima skirti monokloninius antikūnus (infliksimabą ir kt.).
Dažnas kintamasis imuninis trūkumas
Bendras kintamasis imuninis deficitas (CVID) yra sindromų grupė, kuriai būdingas antikūnų sintezės ir ląstelių imuniteto defektas. Patikimas CVID diagnostinis kriterijus yra reikšmingas dviejų ar trijų pagrindinių izotipų imunoglobulinų kiekio sumažėjimas abiejų lyčių atstovams kartu su vienu iš šių požymių:
Ligos pradžia nuo 2 metų amžiaus;
Izohemagliutininų trūkumas ir (arba) silpnas atsakas į vakcinaciją;
Kitų agammaglobulinemijos priežasčių neįtraukimas.
Kai kuriems pacientams CVID vystymosi priežastis yra genų, koduojančių molekules, dalyvaujančias B ląstelių brendimo ir išgyvenimo procesuose, mutacijos: BAFF-R (B ląstelių aktyvuojančio faktoriaus receptorius),Blimp-1 (B limfocitų sukeltas brendimo baltymas-1)ir ICOS (Indukuojamas kostimuliatorius).Pažeidžiamas B-limfocitų gebėjimas diferencijuotis į plazmos ląsteles, išsivysto antikūnų gamybos defektai, galimi T-limfocitų disfunkcijos, pastebimas padidėjęs polinkis į infekcines ligas. Sindromas gali pasireikšti ankstyvoje vaikystėje, paauglystėje ar jaunuose suaugusiuose.
Laboratorinių tyrimų duomenys.IgG ir IgA (maždaug 50% pacientų) ir IgM (iki nenustatomo kiekio) lygis yra žymiai sumažėjęs. B-limfocitų kiekis kraujyje yra normalus arba sumažėjęs. T-limfocitų skaičius daugumoje pacientų yra normalus. Sunkiems pacientams gali išsivystyti limfopenija (mažiau nei 1500x103 ląstelių 1 litre kraujo). NK ląstelių skaičius yra sumažėjęs. Specifinių antikūnų gamyba, reaguojant į imunizaciją, sumažėja arba jo nėra. Labai pakenktas limfocitų dauginimasis ir IL-2 susidarymas veikiant mitogenams ir antigenams.
Klinikinis vaizdas.Aptinkamos pasikartojančios bakterinės infekcinės ligos, lokalizuojančios daugiausia kvėpavimo takus ir paranalinius sinusus. Diagnozės metu kvėpavimo takų infekcijos gali pereiti į bronchektazę ir difuzinius plaučių audinio pažeidimus. Galimas infekcinis virškinimo sistemos pažeidimas, pasireiškiantis viduriavimu, steatorėja ir malabsorbcija (ir, atitinkamai, svorio kritimu). Infekcijos, kurias sukelia Giardia lamblia, Pneumocystis cariniiarba šeimos virusai Herpetoviridae.CVID sergantiems pacientams būdingas pūlingas artritas, kurį sukelia mikoplazmos ir ureaplazmos. Enterovirusinės infekcijos pasireiškimai gali būti encefalomielitas, poliomielitas ir panašūs į dermatomiozitą sindromai, odos ir gleivinės pažeidimai. Autoimuninisligos yra sunkios ir gali nulemti CVID prognozę. Kartais pirmieji klinikiniai CVID pasireiškimai yra artritas, opinis kolitas ir Krono liga, sklerozuojantis cholangitas, malabsorbcija, SLE, nefritas, miozitas, autoimuninė plaučių liga, pasireiškianti limfoidinio intersticinio pneumonito forma, neutropenija,
trombocitopeninė purpura, hemolizinė anemija, žalinga anemija, totalinė alopecija, tinklainės vaskulitas, jautrumas šviesai. CVID sergantiems pacientams žymiai dažniau pasireiškia piktybiniai navikai(15% atvejų), į sarkoidozę panašios granulomos ir nepiktybinis limfoproliferacija. Gydymas.
Antibakterinė chemoterapija.
Pakaitinė terapija: į veną leidžiami imunoglobulino preparatai visą gyvenimą skiriami kas 3-4 savaites.
Autoimuninių komplikacijų atveju - imunosupresinis gydymas (gliukokortikoidai, azatioprinas, ciklosporinas A) ir galbūt skiriami monokloniniai antikūnai (infliksimabas ir kt.).
Hyper-IgM sindromai
Hiper-IgM sindromai yra gana retos ligos, kurioms būdingas ryškus IgG, IgA sumažėjimas arba visiškas nebuvimas ir normali ar padidėjusi IgM koncentracija serume. Taip yra dėl B-limfocitų nesugebėjimo perjungti imunoglobulino klases ir kintamų domenų hipermutagenezės. Iki šiol buvo nustatyti 6 genetiniai defektai, dėl kurių išsivystė hiper-IgM sindromas.
. 1 tipas (HIGM 1).X susietas CD40 ligando trūkumas (70% hiper-IgM sindromų atvejų), dėl kurio T ląstelės nesugeba veiksmingai sąveikauti su B limfocitais.
. 2 tipas (HIGM 2).Autosominis recesyvinis, susijęs su AID defekto sukeltu citidino deaminazės (geno 1) aktyvavimu Aicda, 12p13)- fermentas, dalyvaujantis keičiant imunoglobulinų klases ir hipermutagenezę.
. 3 tipas (HIGM 3).Autosominis recesyvinis, susijęs su CD40 molekulės geno mutacija. Tuo pačiu metu B ląstelės pačios nesugeba veiksmingai sąveikauti su T limfocitais. Fenotipinės apraiškos yra panašios į 1 tipo.
. 4 tipas (4 HIGM).Autosominis recesyvinis; kai kuriais atvejais atsiranda mutacijos de novo.Susijęs su UNG defektu - uracilo-DNR glikozilaze - fermentu, kuris taip pat dalyvauja
keičiant imunoglobulinų klases, tačiau po AID veikimo. Šiuo atveju hipermutagenezė neturi įtakos ir sindromas vyksta mažiau sunkiai.
. 5 tipas (HIGM 5).Tik klasės keitimo defektas, hipermutagenezė neturi įtakos. Priežastinė mutacija dar nebuvo nustatyta, tačiau, matyt, yra fermento, veikiančio po to, defektas
. 6 tipas (HIGM-ED).X susietą, susietą su disidrotine ektodermine displazija, sukelia NEMO (NF-kB moduliatoriaus) trūkumas, dėl kurio sutrinka CD40 signalizacija.
X susietas hiper-IgM sindromasnustatomi dažniau nei kiti. Jis vystosi su CD40L koduojančio geno defektu (CD154, genas yra ant Xq26-q27.2)yra CD40 ligandas. Nepakankamas T-limfocitų CD40L ekspresija lemia tai, kad B-limfocituose esančių imunoglobulinų klases negalima pakeisti IgM nuo kitų izotipų, taip pat pažeidžiamos atminties B-ląstelės, T-ląstelių repertuaras ir Th1-ląstelių atsakas, nukreiptas prieš intraląstelinius mikroorganizmus. Berniukai serga
Laboratorinių tyrimų duomenys.IgG, IgA, IgE nustatyti negalima arba jie nustatomi labai mažais kiekiais. IgM lygis yra normalus (50% atvejų) arba padidėjęs, dažnai reikšmingai. T ir B ląstelių skaičius yra normalus; sumažėjęs T ląstelių proliferacinis atsakas, kurį sukelia antigenai. IgM yra polikloniniai, kartais monokloniniai. Atskleiskite IgM izotipo autoantikūnus (antieritrocitus, antitrombocitus, antitiroidus, antikūnus lygiųjų raumenų audinių antigenams). Limfoidiniame audinyje nėra gemalo centrų, tačiau yra plazmos ląstelių.
Klinikinis vaizdas.Pirmosios apraiškos pasireiškia kūdikystėje ir ankstyvoje vaikystėje. Pakartota infekcijosskirtinga lokalizacija (pirmiausia kvėpavimo takai), įskaitant oportunistinę (sukelta Pneumocystis carinii).Taip pat būdingi virusų pažeidimai (citomegalovirusas ir adenovirusai), Criptococcus neoformans,mikoplazmos ir mikobakterijos. Kriptosporidinė infekcija gali sukelti ūminį ir lėtinį viduriavimą (išsivystantį 50% pacientų) ir sklerozuojantį cholangitą. Anemija, neutropenija, burnos gleivinės išopėjimas, gingivitas, opinis
stemplės pažeidimai, įvairios žarnyno dalys, opinis kolitas. Jie atskleidžia polinkį autoimuniniai sutrikimai(seronegatyvus artritas, glomerulonefritas ir kt.) ir piktybiniai navikai (daugiausia limfoidinis audinys, kepenys ir tulžies takai). Gali išsivystyti limfadenopatija, hepato- ir splenomegalija. Gydymas
Reguliarus pakaitinis gydymas intraveniniu imunoglobulinu.
Antibakterinė chemoterapija. Pneumocistinės pneumonijos profilaktikai ir gydymui naudojami kotrimoksazolas [sulfametoksazolas + trimetoprimas] ir pentamidinas.
Norėdami išvengti kepenų ir tulžies takų pažeidimo, naudokite tik virtą arba filtruotą vandenį, reguliariai atlikite tyrimus (ultragarsu, kepenų biopsija, jei nurodyta).
Gydant neutropeniją ir burnos išopėjimą, naudojami gliukokortikoidai ir granulocitų kolonijas stimuliuojančio faktoriaus preparatai.
Išsivysčius autoimuninėms komplikacijoms, skiriama imunosupresinė terapija (gliukokortikoidai, azatioprinas, ciklosporinas A), taip pat vaistai, pagrįsti monokloniniais antikūnais.
Optimalus gydymas yra kaulų čiulpų transplantacija iš HLA suderinamų donorų (išgyvenamumas 68%, geriausia tai padaryti iki 8 metų).
Tai yra dažniausias pirminis imunodeficitas susitinka 1/300 - 1/700 dažniu. Pirmą kartą buvo ...
aprašė J. Heremansas 1960 m.
Daugeliu atvejų aiškus ligos paveldėjimo vaizdas nepastebimas, retesniais atvejais, kai IgA praranda šeimą, autosominis recesyvinis paveldėjimas įvyksta dažniau nei dominuojantis paveldėjimas. IgA trūkumas kartais siejamas su 18 chromosomos anomalijomis.
IgA nuo kitų imunoglobulinų skiriasi angliavandenių ir sialo rūgščių kiekiu ir galimybe suformuoti dimerius, trimerius ir net tetramerius. Yra 2 IgA molekulių variantai:
- IgA serumas visada yra monomeras;
- sekretorinis IgA - sudaro di-, tri- ir tetramerius prieš tapdamas neatsiejama sekreto (seilių, ašarų, pieno, virškinimo, kvėpavimo ir šlapimo takų sekrecijos) dalimi.
Sekretorinės IgA formavime dalyvauja speciali baltymų grandinė - J grandinė. Todėl tai yra visa baltymų sistema. Taigi - IgA patologijos formos:
- Bendras IgA trūkumas yra susijęs su IgA monomero sintezės sutrikimais. Dėl to sumažėja tiek serumo, tiek sekrecijos IgA kiekis. Pažeidžiama tiek vietinė, tiek bendroji apsauga;
- sekretorinių molekulių susidarymo defektas - SIgA. Priežastis gali būti J-grandinės nebuvimas, dėl kurio pažeidžiamas vietinis imunitetas;
- vyraujantis IgA serumo sintezės pažeidimas, kai išsiskiria tik sekrecinės IgA formos, o serumo IgA monomerai praktiškai neišskiriami.
Klinikiniai simptomai priklauso nuo igA trūkumas ir gali pasireikšti atskirų organų ir sistemų patologija. Dažniausiai pasireiškia infekciniai gleivinės pažeidimai.
Iš virškinamojo trakto - tai lėtinis gastritas, opinis ir hemoraginis kolitas, ileitas, stomatitas. Iš kvėpavimo sistemos pusės - sloga, sinusitas, bronchitas, kurie greitai tampa lėtiniai. Dažnai pasitaiko lėtinė bronchopneumonija, pasireiškianti bronchektaze ir plaučių abscesais. Taip pat buvo nustatytas ryšys tarp sumažėjusio sekrecinio IgA kiekio ir padidėjusio atopinės alergijos dažnio bei autoimuninių ligų (sisteminės raudonosios vilkligės, reumatoidinio artrito, autoimuninio tiroidito) išsivystymo.
Asmenys, turintys selektyvų daugiausia serumo monomerinio IgA trūkumą, neturi klinikinių imunodeficito pasireiškimų.
Laboratoriniai tyrimai:rodo, kad tokių pacientų bendras T ir B-limfocitų skaičius periferiniame kraujyje yra normos ribose. Serumo ir (arba) sekrecijos IgA kiekis yra smarkiai sumažėjęs, o IgM ir IgG lygis yra normos ribose. Kai kuriems pacientams (turintiems atopinės alergijos pasireiškimų) IgE lygis gali būti padidėjęs.
Gydymas: simptominis. Naudojant pakaitinę imunoglobulino terapiją reikia būti atsargiems, nes vartojant pakartotinai, kyla anafilaksinio šoko rizika, nes padidėja IgG ir IgM klasių antiimunoglobulinų gamyba.
Prognozė: ligos eiga dažniausiai būna gerybinė.
Diagnostinis kriterijus - vyresnių nei 4 metų pacientų serumo IgA koncentracijos sumažėjimas mažesnis nei 0,07 g / l, esant normalioms IgG ir IgM koncentracijoms, ir neįtraukiant kitų hipogammaglobulinemijos priežasčių. Diagnostiškai reikšminga:
Izoliuotas IgA koncentracijos serume sumažėjimas (mažiau nei 0,05 g / l) su normaliu kitų imunoglobulino izotipų kiekiu vyresniems nei 1 metų vaikams, IgAl ir IgA2 trūkumas. IgM ir IgG kiekis yra normalus. Tačiau kai kuriems pacientams yra IgG2 trūkumas;
- jei IgA lygis yra nuo 0,05 g / l iki 0,2 g / l, tada diagnozuojamas dalinis IgA trūkumas; normalus T-limfocitų ir jų poklasių skaičius;
- paprastai normalus B-limfocitų skaičius (CD19 \\ CD20);
- normalus NK ląstelių skaičius (CD16 CD56).
Pacientams, turintiems IgA trūkumą, ypač jei nėra sekrecijos IgA, būtina ištirti IgA poklasių lygį. Kai kuriems pacientams selektyvus IgA trūkumas ateityje gali progresuoti plėtojant CVID. Būtina reguliariai stebėti ilgalaikį imunoglobulinų kiekį (įskaitant besimptomius pacientus).
Autoantikūnų (antinuklearinių, antitiroidinių ir kt.) Nustatymas.
Su maistu nepakantumas arba malabsorbcijai reikalingi alergijos tyrimai ir antikūnų prieš pieną bei antigluteno IgG nustatymas.
Gydymas... Pacientams, kuriems yra besimptomis selektyvus IgA trūkumas, nuolatinio gydymo nereikia. Pacientams, turintiems infekcinių ligų pasireiškimų, profilaktikos tikslais skiriami antibiotikai. Infekcinės ligos metu visiems pacientams atliekamas intensyvus antibakterinis gydymas. Įprastinė imunizacija pacientams nėra draudžiama. Imunoglobulino pakaitinis gydymas yra draudžiamas, kai pacientui nustatomi anti-IgA autoantikūnai. Reikėtų nepamiršti, kad selektyvus IgA trūkumas reiškia neištaisytus pirminius imuninius defektus. Terapinės priemonės sumažėja iki simptominės infekcinių, alerginių ir autoimuninių ligų terapijos. Imunotropiniai vaistai skiriami daugiausia dėl padidėjusio infekcinio sergamumo pasireiškimo.
Prognozė... Pacientų, sergančių selektyviu IgA trūkumu, prognozė priklauso nuo to, ar yra kartu specifinių antikūnų, alergijų ar autoimuninių ligų defektų. Dažnai besimptomė ligos eiga gali būti sutrikdyta dėl išorinių žalingų veiksnių poveikio, pavyzdžiui, esant stresinei situacijai, taikant imunosupresiją, chemoterapiją ir kt.