It should be differentiated from the real syndrome of hypervitaminosis D, in which, as a result of enzymatic deficiency of the renal tubular apparatus, severe metabolic disorders develop in the child’s body. V. V. Shitskova et al. (1971), based on their own observations, provide the following differential diagnostic table of these diseases (Table 7) with some of our additions.
| Indicators | Hypervitaminosis D | De Toni-Debreu-Fanconi syndrome |
| Frequency | Relatively often | Rarely |
| Pathogenesis | Disturbance of metabolic processes, mainly calcium, due to an overdose of vitamin D | Enzymopathy. Congenital tubulopathy. Impaired reabsorption of phosphorus, glucose and amino nitrogen |
| Clinical picture | Dry and pale skin, thirst, vomiting, constipation, malnutrition, hypertension, liver enlargement | Dry and pale skin, anorexia, thirst, vomiting, constipation, polyuria, malnutrition, liver enlargement. No hypertension. Hypotonia of muscles |
| Biochemical blood tests | Hypercalcemia in acute period. Phosphorus is low. Sugar and protein are normal. Alkaline phosphatase is not changed | Calcium is normal or reduced. Phosphorus is sharply reduced. Sugar and protein are reduced, alkaline phosphatase activity is sharply increased. Metabolic acidosis |
| Urine | Sulkowicz's reaction is positive. Proteinuria, microhematuria, leukocyturia. Amino nitrogen sugar is often normal | Sulkowicz's reaction is negative. Proteinuria, phosphaturia, glucosuria, aminoaciduria |
| X-ray of tubular bones | Expansion and compaction of pre-calcification zones | Osteoporosis of long bones, calcification zones are poor |
Fanconi syndrome- amine diabetes. Cystine metabolism disorder accompanied by glycosuria, aminoacidouria, phosphaturia, increased secretion alanine, loss of alkalis. Caused by a hereditary defect of the renal tubules, causing the inability to reabsorb glucose, amino acids and phosphate. The metabolism of cystine is disrupted, which is deposited in the form of crystals in the reticuloendothelial system, cornea, renal tubules and other tissues. This leads to progressive failure over time various functions renal tubules.
Fanconi syndrome should be distinguished from cystinuria, in which cystine is not deposited in the tissues. With the syndrome, the functional ability of the glomerular apparatus remains normal. The serum calcium concentration is normal, the level of inorganic phosphorus decreases sharply. The radiograph shows osteomalacia and diffuse depletion of bones in alkali.
De Toni-Debreu-Fanconi syndrome (disease) is one of the rickets-like diseases.
Story
The renal syndrome was identified by the Swiss pediatrician Fanconi among those previously described by other researchers individual parts diseases. In 1931, he described glucosuria and albuminuria in a child with dwarfism and rickets; 2 years later, de Toni added clinical picture hypophosphatemia, and soon Debray described aminoaciduria.
Etiopathogenesis
The type of inheritance is autosomal recessive; an autosomal dominant form with the gene localization on chromosome 15q15.3 has also been identified. The expressivity of the mutant gene in the homozygous state varies significantly. There are sporadic cases caused by a fresh mutation. It is believed that the disease is based on genetically determined defects in enzymatic phosphorylation in the renal tubules (combined tubulopathy), deficiency of enzymes of the 2nd and 3rd complexes of the respiratory chain - succinate dehydrogenase and cytochrome oxidase. A number of authors classify this disease as a mitochondrial disease.
The reasons described above lead to disruption of the processes of energy supply for the transport of phosphates, glucose and amino acids in the renal tubules and their increased excretion in the urine, as well as to disruption of the mechanisms of maintaining acid-base balance. Developing metabolic acidosis and lack of phosphorus compounds contribute to disruption of the formation bone tissue according to the type of osteomalacia and rickets-like changes in the skeleton.
Clinical picture
The first signs appear in the second half of the first year of life, a detailed symptom complex is formed by the second year of life. Sometimes there is a manifestation at 6-7 years. Initial signs- thirst, polyuria, vomiting, sometimes prolonged low-grade fever. In the second year, a lag is detected physical development and bone deformities of the lower extremities (valgus or varus), chest, forearms and humerus, decreased muscle tone. Radiologically, systemic osteoporosis of varying degrees of severity, thinning of the cortical layer of tubular bones, loosening of growth zones, and a lag in the growth rate of bone tissue from the biological age of the child are determined. Bones become brittle.
Depending on severity clinical manifestations and metabolic disorders, there are two clinical and biochemical variants of the disease. First is characterized by a significant delay in physical development, a severe course of the disease with severe bone deformations and often bone fractures, severe hypocalcemia (1.6-1.8 mmol/l), and a decrease in calcium absorption in the intestine. At second In this variant, a moderate delay in physical development, a mild course with minor bone deformations, normocalcemia and normal absorption of calcium in the intestine are noted.
Biochemical disorders
- decreased calcium levels in the blood;
- decreased phosphorus levels in the blood;
- increased alkaline phosphatase levels;
- development of metabolic acidosis (pH = 7.35-7.25; BE = -10...-12 mmol/l) due to a defect in the reabsorption of bicarbonates in the proximal tubules;
- normal urinary calcium excretion;
- increased clearance of urinary phosphates, absorption of phosphates in the intestine is not affected;
- development of glucosuria (20-30 g/l and above);
- development of generalized hyperaminoaciduria;
- dysfunction of ammonia acidogenesis - decreased titration acidity, increased urine pH above 6.0;
- development of hypokalemia.
Differential diagnosis
De Toni-Debreu-Fanconi disease must be differentiated from the secondary syndrome found in other hereditary and acquired diseases (
Fanconi syndrome (full name - de Toni-Debreu-Fanconi) is a congenital pathology, which is expressed in serious dysfunction of the proximal renal tubules, namely, a violation of the secondary absorption (absorption into the blood) of substances filtered by the kidneys, which leads to glucosuria ( high sugar in the urine), phosphaturia (impaired metabolism of phosphorus and calcium), aminoaciduria (increased excretion of amino acids in the urine) and a decrease in the concentration of bicarbonates that regulate blood acidity.
De-Toni-Debreu-Fanconi syndrome
Fanconi syndrome is very rare disease, mainly found in children, and medical statistics its frequency corresponds to 1 sick baby per 350 thousand newborns of both sexes.
It is observed extremely rarely in adults, developing against the background of acquired pathologies. Pathology code according to ICD-10: E72.O.
Causes
The nature and causes of the genetic defect Fanconi syndrome are not well understood today.
It is assumed that the pathology is based on either defects in the transport proteins of the renal tubules or a gene mutation that distorts the function of enzymes that regulate the reabsorption of glucose, amino acids and phosphorus.
There is research data on point defects in mitochondria leading to improper functioning of the kidney tubules.
The disease is also associated with fructose intolerance, chronic poisoning toxins (heavy metals, ifosfamide, aminoglycosides), vitamin D deficiency, amyloidosis, deficiency of a number of cellular enzymes (pyruvate carboxylase, phosphoenolpyruvate carboxykinase and others), tyrosinemia, metachromatic leukodystrophy, galactosemia, cystinosis, glycogenosis.
According to other experts, Fanconi syndrome may be an isolated pathology - namely, one of severe forms rickets-like pathologies that are hereditary in nature.
Studies confirm that in Fanconi syndrome, cellular energy metabolism with the participation of ATP (adenosine triphosphate) and intercellular transport in the tubules of the main element of the kidney - the nephron.
Due to insufficient enzyme functions, losses of glucose, phosphates, and amino acids occur, and the kidney tubules experience energy deficiency. In this case, important substances leave in the urine, leading to degenerative changes in bone tissue - rickets.
Since medicine has not yet come to a clear conclusion about the causes of Faconi syndrome, this condition is also designated by other terms: “glucophosphamine diabetes”, “idiopathic renal Fanconi syndrome”, “D-resistant rickets”, “renal dwarfism with D-resistant rickets” , " hereditary syndrome Fanconi."
Forms and pathogenesis
There are two types of Fanconi syndrome:
- hereditary (congenital, idiopathic), which refers to the primary form;
- acquired, which is considered as a secondary form of the disease.
Experts associate the hereditary (genetic) type with a defect in the X chromosome, which is inherited in a dominant and recessive manner, so genetic prediction of its manifestation in future offspring is not an easy task. If the pathology is of the congenital (primary form) type, then it is detected in the child in the infancy period up to one year. Therefore, the primary form is called “infant”.
Degree gene mutation also determines the severity of the syndrome itself. Thus, hereditary complete Fanconi syndrome manifests itself in the presence of 3 basic biochemical defects, which include glucosuria, aminoaciduria, phosphaturia, incomplete - in two of them.
Typically, a genetically determined pathology is accompanied by other congenital ailments: cystinosis, Wilson syndrome, Dent syndrome, Lowe syndrome, fructose intolerance, tyrosinemia (the body’s inability to effectively break down the amino acid tyrosine), galactosemia (dysfunction of converting sugar into glucose), and excessive accumulation of glycogen.
Development of Fanconi syndrome
Acquired syndrome (secondary), unlike congenital, is not accompanied, but is a consequence of existing pathologies:
- tyrosinemia type I;
- cystinosis (impaired metabolism of the amino acid cystine with subsequent kidney damage);
- fructose intolerance;
- Wilson-Konovalov disease;
- galactosemia;
- glycogenosis (abnormal accumulation of glycogen in tissues and organs) type XI;
- hereditary kidney pathologies;
- (disturbance of protein metabolism leading to sclerosis, atrophy, organ dysfunction);
- tubulointerstitial (nephritis with damage to tissue, kidney tubules);
- hyperparathyroidism ( endocrine disease, in which the normal levels of calcium and phosphorus in the blood are disrupted);
- malignancies: myeloma, lung, pancreatic, lung, light chain disease, ;
- deep burns.
In addition, the syndrome can be provoked by the following conditions:
- organ transplantation with low tissue compatibility;
- vitamin D deficiency;
- poisoning with uranium, bismuth, mercury, lead, cadmium;
- contact with toluene, maleic acid, lysol;
- use of nephrotoxic pharmacological agents, such as: Gentamicin, platinum drugs, expired tetracycline-based drugs, Didanosine, Cidofovir, anti-cancer chemotherapy drugs - Ifosfamide, Streptozocin.
Symptoms and signs
In hereditary (congenital) form
Primary symptoms appear in the first months of life, rarely after one and a half years.
First of all, the following conditions are noticed in a newborn baby:
- frequent urination (polyuria);
- increased thirst (polydipsia);
- prolonged constipation;
- frequent attacks of causeless vomiting;
- asthenia (general fatigue), muscle weakness;
- unexplained “jumps” in temperature up to 37.5 – 38 C;
- bloated tummy.
As a rule, during the period when attacks of vomiting begin and the baby’s temperature rises, the baby is taken to the pediatrician. An experienced specialist must determine that the combination of signs that concern parents is not related to acute respiratory infections, acute respiratory viral infections or enterovirus infection.
A competent pediatrician is able to recognize Fanconi syndrome in a timely manner. A laboratory research– confirm suspicions if three (or two) basic signs are detected: glucosuria, generalized hyperaminoaciduria and hyperphosphaturia, characteristic of this pathology.
After mild and rather vague symptoms in the next year and a half, the symptoms characteristic of Fanconi syndrome are clearly recorded:
- Early dwarfism (short stature), caused by the constant removal from the body of the most important amino acids, glucose, calcium, and phosphates. The first six months of normal height and weight are replaced by a deficit of body weight (up to 30%) and height (from 2 to 21%).
- Rickets, caused by massive excretion of calcium and phosphates, becomes noticeable after 10–12 months of life, and has features characteristic of Fanconi syndrome: the baby’s head is usually slightly deformed, but the large bones of the legs and arms show significant curvatures - varus-type deformities when the baby’s lower legs are curved like a “wheel”, or valgus (shaped like the letter “X”). The bones of the chest and spine are also bent.
- Delay in mental and physical development.
- Unsociability, timidity, complexness.
- Polydipsia and polyuria can progress and regress without going away completely.
- Moderate muscle hypotonia, expressed in slowness, difficulty in movements, leading to the fact that children 5–6 years old are unable to walk.
- Bone pain moderate intensity that prevent the child from walking. They are more pronounced at the level of the legs, pelvis and spinal column. The gait, if the child walks, becomes “duck-like” and uncertain.
- High probability of tubular bone fractures due to mineral deficiency in bone tissue.
- Osteomalacia or softening of bones due to the destruction of bone tissue as a result of a lack of calcium and phosphorus salts.
- Reduced immune defense to infections, which manifests itself in frequent viral diseases, otitis, pneumonia.
- Paralysis caused by lack of potassium.
- Ophthalmological pathologies, such as retinitis pigmentosa, congenital cataracts.
- Development of pathologies nervous system, ENT (ear, nose, pharynx, larynx) and gastrointestinal tract, of cardio-vascular system, anatomical abnormalities of the urinary system due to massive metabolic disorders.
- In isolated cases - endocrine disorders
With the progression of tubular disorders (impaired transport organic matter, minerals, electrolytes) by the age of 10–12 years, children have an increased likelihood of developing chronic renal failure, which threatens his life.
Visible symptoms of Fanconi syndrome in children 
In adult patients with the development of a secondary form
If acquired Fanconi syndrome develops in adults with other diseases or pathological conditions, then its manifestations are often combined with manifestations of the provoking disease.
However, basic signs have been identified:
- Increased volume of urine per day (up to 2 liters or more) and acute thirst, also characteristic of patients of early childhood.
- General and muscle weakness, bone pain.
- High probability of persistent increase in blood pressure () due to kidney dysfunction.
- Osteomalacia (bone destruction).
- Acidosis (increased blood acidity) due to retention of oxidation products in the body, leading to hypokalemia (potassium deficiency).
- Nephrocalcinosis is a high deposition of calcium salts in the kidneys with fever, chills, nausea, severe pain in the abdomen, groin, and ovaries characteristic of this condition.
- Hypokalemia (low potassium intake) causing severe heart complications including life threatening arrhythmias.
- Rapid (in the absence of treatment) formation chronic failure kidney
Among women
The most unfavorable course of Fanconi syndrome occurs when it develops in postmenopausal women. At this time, against the background of decreased hormone production, a natural decrease in bone density (osteopenia) occurs.
When this condition is combined with increased bone fragility due to lack of minerals, there is a high likelihood of severe compression fractures vertebrae, femoral neck and subsequent disability.
Diagnostics
To confirm or refute the diagnosis, bone examination and in-depth biochemical studies of blood and urine are performed using x-rays.
Laboratory
Detectable changes in the biochemistry of urine and blood:
| Signs | Indicators |
|---|---|
| low calcium and phosphorus content | less than 2.1 mmol/l and 0.9 mmol/l, respectively |
| acidosis (“acidification” of the blood) | BE = 10 – 12 mmol/l |
| glucosuria (increased sugar in urine) | 2 – 3% and above |
| hyperaminociduria (excretion of important amino acids alanine, arginine, glycine, prolit with urine) | up to 2 – 2.5 g/day |
| excretion of calcium in urine | 1.5 – 3.5 mmol/day |
| increased urine pH (acidity) due to abnormally high loss of bicarbonates | up to 6.0 |
| increase in relative density of urine | 1,025 – 1,035 |
The examination also reveals:
- increased alkaline phosphatase activity;
- excessive excretion of sodium and potassium salts from the body;
- proteinuria (the appearance of protein in the urine) in the presence of light chains of immunoglobulins, lysozyme, low molecular weight proteins, beta 2-microglobulins;
- increased clearance (filtration rate) uric acid with its reduced serum content
- decrease in the activity of energy exchange enzymes: succinate dehydrogenase, a-glycerophosphate dehydrogenase, glutamate dehydrogenase;
- an increase in the amount of lactic and pyruvic acid in the blood.
Instrumental
Diagnosis of Fanconi syndrome requires the mandatory use of bone radiography to identify deformities of the skeleton, limbs, detect signs of osteomalacia, osteoporosis, and in children, additionally, delayed bone growth in comparison with the norm for calendar age.
The following anomalies in bone tissue are observed:
- coarse fibrous, cellular structure with lacunae, weak mineralization, abnormal growths in the form of “spikes” in the femur and tibia;
- signs of epiphysiolysis (partial or complete cessation of bone growth in length in an immature skeleton, leading to asymmetry of the limbs);
- fractures of tubular bones (at a late stage) against the background of the development of osteoporosis, the degree of which is determined using x-ray densitometry;
- accumulation of the radioisotope in areas of intense bone growth.
In the kidneys:
An electronic examination of a kidney tissue biopsy (biopsy) reveals a characteristic change in the shape of the tubules in the form of a “swan neck”, thinning, atrophy (decrease in volume) of the epithelial tissue in the presence of an increased number of mitochondria in it, fibrosis (abnormal growth) of the connective tissue.
Required research:
Differential
Pathology should be distinguished from all isolated diseases that can provoke the occurrence of acquired Fanconi syndrome, acquired unhealthy conditions and intoxications.
In addition, in infants, the pediatrician must differentiate the state of acute deficiency of vitamin D in Fanconi syndrome from its excess due to the use of artificial supplements or calcium metabolism disorder.
Differences in hypervitaminosis D and de Fanconi syndrome in children under one year of age:
| Options | Hypervitaminosis D | De Fanconi syndrome |
|---|---|---|
| Frequency | Often | Rarely |
| Symptoms (similar) | Dry skin, pallor, acute thirst, vomiting, prolonged constipation, weight and height deficiency, liver enlargement | |
| Symptoms (varies) | Hypertension (frequent rises in blood pressure) | Exhaustion, polyuria, muscle hypotonia, high blood pressure no blood |
| Blood | Excess calcium in the acute period. Reduced phosphorus content. Alkaline phosphatase, sugar, protein - normal | Calcium is often normal (may be low). Glucose and protein levels are reduced. Phosphorus is sharply reduced. The activity of alkaline phosphatase is sharply increased - 2–3 times. Increased blood acidity |
| Urine | The Sulkowicz calcium excretion test is positive. The presence of protein, blood (more than 2 - 3 red blood cells in the field of view), leukocytes. Amino nitrogen sugar is usually normal | The Sulkowicz test is negative. Protein, phosphates, sugar elevated, aminoaciduria |
| Bones | Calcification zones are expanded and compacted | Osteoporosis of long bones, calcium deficiency in areas of calcification |
Specialists dealing with Fanconi syndrome and its complications: nephrologist, orthopedist, hematologist, endocrinologist, urologist, ophthalmologist, geneticist.
Treatment
Rational, well-designed therapy can reduce the impact on the brain, skeletal system and organs of excessive loss of essential amino acids, minerals, glucose and protein excreted in urine.
Therefore, treatment for the syndrome is aimed at the following tasks:
- Maximum possible correction deficiency of potassium, bicarbonates, changes in acid-base balance blood to reduce acidosis.
- Therapy of phosphate-diabetes (D-resistant rickets) with an emphasis on the inadmissibility of fluid restriction.
- Treatment of the underlying disease that provokes the development of the acquired syndrome in adults.
Medication
 To mitigate the loss of phosphorus and calcium, use special drugs with vitamin D, these are l,25(OH)D3 and l(OH)D3.
To mitigate the loss of phosphorus and calcium, use special drugs with vitamin D, these are l,25(OH)D3 and l(OH)D3.
Initial doses of vitamin D3 per day are 10 - 15 thousand IU. The dose is increased gradually, increasing it every 12 to 14 days (under the control of the Sulkovich test and phosphorus levels in the blood). In the absence of signs of intoxication and small excretion of calcium in the urine, it is allowed to increase the dose, bringing it to 100 - 150 thousand IU per day, and continue therapy until normal indicators phosphorus and alkaline phosphatase in the blood. When their values stabilize, the dose should not be increased further.
Vitamin D therapy is carried out in several courses to prevent crises of progression of rachitic deformities in the bones.
The best option is the use of active metabolites D3 - Oksidevit (0.5 - 1.5 mcg per day), calciotriol (Rocaltrol).
Include calcium supplements (calcium gluconate per day up to 1.5 - 2 grams), phosphorus (0.5 - 1 gram per day), phytin.
Of inorganic phosphates, use Albright's mixture, taking 1 large spoon 4 to 5 times a day. Phosphates are used in the form of a solution and tablets in doses calculated at 10 mg per kilogram of weight, 4 times a day (always with vitamin D preparations to avoid hyperparathyroidism).
In cases of severe potassium deficiency, Panangin and Asparkam are used.
For any prescription, the CBS or acid-base status of the blood is constantly monitored. Normally, blood is slightly alkaline, with a pH in the range of 7.35 – 7.45. In acidosis, when the pH value drops below 7.35, the blood becomes highly acidic.
In these cases, intravenous infusion of 4% sodium bicarbonate solution or drinking a solution (50 - 60 ml per day), which includes lemon acid- 2 grams, sodium citrate - 3 g, potassium citrate - 3.3 g, water 100 ml. 1 ml of such an alkalizing mixture contains 1 mmol of sodium and potassium. High acidity of the blood can also be neutralized with baking soda (sodium bicarbonate).
Some experts, based on practical results, recommend Unithiol as a means of increasing the activity of thiol-dependent enzymes.
For cystinosis, the following is prescribed: Dithiotrental at the rate of 25 mg per kilogram of the patient’s weight after 3 hours; Cysteamine in daily dose at the rate of 90 mg/kg.
Available positive results the use of Penicillamine, which lowers the concentration of pyruvic acid in the blood, reduces the degree of excretion of amino acids and promotes the growth of alkali reserves in the body.
Hormonal anabolics, including Methyltestosterone, have a good effect on the functioning of the kidney tubules.
Treatment in a hospital is indicated for obvious metabolic disorders, including hypo- and hyperglycemia, and skeletal deformities.
Prevention
Modern prevention congenital syndrome Fanconi, in the presence of a similar pathology in the family, involves preliminary genetic counseling. The risk of developing the disease for so-called siblings, that is, sisters and brothers, is about 25%.
In the secondary form of the syndrome, its symptoms decrease or completely disappear with active treatment of the underlying disease or acquired pathological condition.
Forecast
The prognosis of the disease depends on the form (primary, secondary) of the syndrome, the severity of the manifestations, and the start of treatment.
For example, the symptoms of an acquired syndrome disappear when the provoking cause is eliminated.
At congenital pathology against the background of the absence of cystine deposits in the tissues, the course of the disease does not pose a serious threat to life; otherwise, especially without appropriate treatment, the patient’s death is predicted up to 10–20 years from increasing kidney failure.
However, even with severe changes in the kidneys: pyelonephritis, tubulointerstitial nephritis, renal failure, with early drug exposure to pathological process and long-term therapy, a certain balance in homeostasis is established, in which the prognosis for a normal quality of life for decades is good.
IN medical practice There are case histories where in children aged 7–8 years, hereditary Fanconi syndrome was practically “stopped” with the onset of long-term remission, a clear improvement in the child’s condition and even recovery.
De Toni-Debrau-Fanconi syndrome is a severe congenital disease, characterized by a variety of children most often suffer from it in the first year of life. Typically found in combination with others hereditary pathologies, but can also manifest as an independent syndrome.
A brief excursion into history
The disease was discovered and studied in 1931 by Dr. Fanconi from Switzerland. Having examined a child with rickets, short stature and changes in urine tests, he came to the conclusion that this combination of signs should be considered as a separate pathology. Two years later, de Toni made his own amendments, adding hypophosphatemia to the existing description, and after some time Debray identified aminoaciduria in such patients.
In the domestic literature, this condition is called the terms “hereditary de Toni-Debreu-Fanconi syndrome” and “glucoaminophosphate diabetes”. Abroad, it is more often called renal Fanconi syndrome.
Reasons for the development of Fanconi syndrome
At the moment, it has not been possible to fully understand what lies at the basis of this serious illness. Fanconi syndrome is believed to be Experts believe that the development of this pathology is associated with a point mutation that leads to malfunction of the kidneys. Numerous studies have confirmed that cellular metabolism is disrupted in the body. It is possible that adenosine triphosphate (ATP), a compound that plays an important role in energy metabolism, is involved. As a result of incorrect enzyme operation, glucose, amino acids, phosphates and other equally useful substances are lost. In such harsh conditions, the kidney tubules do not receive the energy they need to function. Useful material excreted along with urine, disrupted metabolic processes, rickets-like changes in bone tissue develop.

Fanconi syndrome is much more common in children than in adults. According to statistics, the frequency of pathology is 1:350,000 newborns. Both boys and girls are affected in equal proportions.
Signs of Fanconi syndrome
The disease can develop at any age, but most often it occurs in children in the first year of life. Glucosuria, generalized hyperaminoaciduria and hyperphosphaturia - this triad of symptoms characterizes Fanconi syndrome. Symptoms develop quite early. First of all, parents notice that their child begins to urinate more often and is constantly thirsty. Babies, of course, cannot say this in words, but from their capricious behavior and constant hanging on the chest or bottle, it becomes obvious that something is wrong with the child.
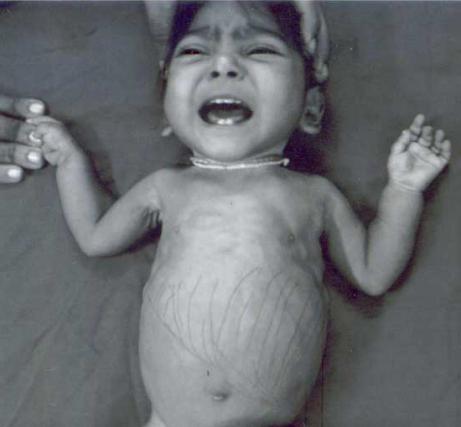
IN further parents Frequent causeless vomiting, prolonged constipation and unexplainable vomiting bring a lot of concern. As a rule, at this stage the child finally gets an appointment with a doctor. An experienced pediatrician may suspect that this combination of symptoms is not at all similar to a common cold. If the doctor turns out to be competent, he will be able to recognize Fanconi syndrome in time.
However, the symptoms do not disappear. Added to these are a noticeable lag in physical and mental development, pronounced curvatures of large bones appear. Typically changes only affect lower limbs, leading to deformation of the varus or valgus type. In the first case, the child’s legs will be bent like a wheel, in the second - in the form of the letter “X”. Both options, of course, are unfavorable for the child’s future life.
Fanconi syndrome in children often includes osteoporosis (premature bone loss) as well as significant growth retardation. Long fractures and paralysis are not excluded. Even if until now the parents have not worried about the baby’s condition, at this stage they will definitely not refuse qualified help.

Fanconi syndrome is quite rare in adults. The whole point is that this serious illness naturally leads to the development of renal failure. In this situation, it is impossible to give any unambiguous prognosis and guarantee a great deal. The literature describes cases when, at the age of 7-8 years, Fanconi syndrome lost its ground, and there was a noticeable improvement in the child’s condition and even recovery. Unfortunately, such options are rare enough in modern practice for any serious conclusions to be drawn.
Diagnosis of Fanconi syndrome
In addition to collecting anamnesis and a thorough examination, the doctor will definitely prescribe some examinations to confirm this disease. Fanconi syndrome inevitably leads to impaired kidney function, which means a routine urine test will be required. Of course, this is not enough to identify all the features of the course of the disease. It is necessary to look not only at the content of protein and leukocytes in the urine, but also to try to detect lysozyme, immunoglobulins and other substances. The analysis will also necessarily reveal high content sugar (glucosuria), phosphates (phosphaturia), significant losses of substances important for the body will be visible. Such an examination can be carried out both on an outpatient basis and in a hospital setting.

Some changes are also inevitable in blood tests. At biochemical research There is a decrease in almost all significant microelements (primarily calcium and phosphorus). A pronounced interference develops normal operation the whole body.
A skeletal x-ray will show osteoporosis (destruction of bone tissue) and deformation of the limbs. In most cases, a lag in bone growth rates and their inconsistency with biological age are detected. If necessary, the doctor may prescribe an ultrasound of the kidneys and other internal organs, as well as examination by related specialists.
Differential diagnosis
There are cases when some other diseases are disguised as Fanconi syndrome. Stands in front of the doctor difficult task to figure out what is really happening to the little patient. Glucoaminophosphate diabetes is sometimes confused with chronic pyelonephritis and other kidney diseases. Changes in urine tests, as well as characteristics bone tissue lesions will help the pediatrician make the correct diagnosis.
Treatment of Fanconi syndrome
It is worth considering the fact that this pathology is chronic. It is quite difficult to completely get rid of unpleasant symptoms; you can only reduce the manifestations of the disease for a while. What does it offer? modern medicine as help for sick children?
Diet comes first. Patients are advised to limit their intake of salt, as well as all spicy and smoked foods. Milk and various fruit sweet juices are added to the diet. Don't forget about (prunes, dried apricots and raisins). In cases where the deficiency of microelements has reached a critical stage, doctors prescribe special vitamin complexes.
Along with the diet, large doses of vitamin D are administered. The patient's condition is constantly monitored - he has to donate blood and urine for tests from time to time. This is necessary in order to identify incipient hypervitaminosis in time and reduce the dose of vitamin D. Treatment is long-term, in long courses, with breaks. In most cases, such therapy helps restore impaired metabolism and prevent serious complications.

If the disease has progressed far, the patient falls into the hands of surgeons. Experienced orthopedists will be able to correct bone deformities and significantly improve the child’s standard of living. Such operations are performed only in case of stable and long-term remission: at least one and a half years.
Forecast
Unfortunately, the prognosis for such patients is unfavorable. In most cases, the disease progresses slowly, sooner or later leading to kidney failure. Deformations of the skeletal bones inevitably lead to disability and a deterioration in the overall quality of life.
Is it possible to avoid this pathology? Undoubtedly, a similar question worries everyone who has encountered Fanconi syndrome. Parents are trying to understand what they did wrong and where they did not keep an eye on their child. It is equally important to know whether the situation threatens to be repeated with other children. Unfortunately, preventive measures have not yet been developed. Couples planning to have another child should consult a geneticist to obtain more complete information about the problem that concerns them.
Wissler-Fanconi syndrome (allergic subsepsis)
This disease has been described only in children aged 4 to 12 years. The cause of this serious pathology is still unknown. It can be assumed that this syndrome is typical autoimmune disease, a special form of rheumatoid arthritis. It always begins acutely, with a rise in temperature, which can stay at 39 degrees for weeks. In all cases, a polymorphic rash appears on the extremities, sometimes on the face, chest or abdomen. Recovery usually occurs without any serious complications. However, some young patients develop severe joint damage over time, leading to disability.
Fanconi syndrome (more correctly de Toni-Debreu-Fanconi syndrome) is a generalized dysfunction of the proximal tubules, which consists of the following disorders (including those described above): 1) proximal tubular acidosis with bicarbonaturia; 2) renal glucosuria; 3) phosphaturia; hypophosphatemia; hypophosphatemic rickets; 4) hyposthenuria (polyuria); 5) aminoaciduria; 6) tubular type proteinuria (immunoglobulin light chains, low molecular weight proteins - p2 microglobulin). In addition, there is a loss of sodium, potassium, calcium, an increase in the clearance of uric acid with a decrease in its content in the serum.
Causes of Fanconi syndrome
Fanconi syndrome can be a primary disease (hereditary or acquired), more often it is secondary, developing in a number of common diseases. The cause of Fanconi syndrome may be hereditary metabolic disorders (cystinosis, galactosemia, Wilson-Konovalov disease); poisoning toxic substances(eg salicylates, expired tetracycline) and heavy metals (lead, cadmium, bismuth, mercury); malignant neoplasms(myeloma, light chain disease, ovarian, liver, lung, pancreatic cancer); lymphogranulomatosis. Fanconi syndrome can also develop with certain kidney diseases, with hyperparathyroidism, paroxysmal nocturnal hemoglobinuria, severe burns.
Clinical signs of Fanconi syndrome
Clinical signs include bone damage (skeletal deformation, bone pain, fractures, diffuse osteomalacia); children develop rickets and growth retardation. There may be polyuria, thirst, rarely muscle weakness (up to paralysis) associated with hypokalemia, hypocalcemic convulsions. Children have reduced resistance to infections. Clinical signs may be absent; the diagnosis in such cases is made on the basis of laboratory data revealing a complex disorder of tubular functions.
Despite the fact that with hereditary forms the first signs appear in childhood, the disease is sometimes recognized at an older age. There are no clinical or laboratory signs that distinguish primary Fanconi syndrome from secondary, so a thorough etiological search should be carried out in each case.
Treatment is carried out with large doses of bicarbonates, citrate mixtures, vitamin D, potassium supplements are prescribed, and a potato-cabbage diet is indicated.

